
The role of circadian rhythm in breast cancer
Introduction
For most organisms, the circadian clock regulates most of the body’s physiological and behavioral functions. The secretion of many endocrine hormones is also regulated by the light/dark cycle. Reciprocally, levels of circulating hormones influence circadian rhythm. This bidirectional information flow is very important for normal physiology, while additional research has shown that the disruption of the circadian rhythm is closely related with malignant growth of the endocrine system such as breast cancer. A remarkable example is that overnight-shift workers tend to have an increased incidence of hormone-related breast cancer as well as other types of malignancies (1,2). Recently, molecular research data has shown that cultured human mammary epithelial cells maintain an inner circadian oscillator, with key clock genes oscillating in a rhythmic fashion. This inner circadian clock is disrupted in both estrogen receptorα-positive (ERα-positive) type or ERα-negative type breast cancer (3,4). Previous investigations demonstrated that the disruption of the circadian rhythm is closely related with breast cancer development.
Mechanism of the mammalian circadian rhythm
The body’s circadian system proficiently coordinates the physiology of living organisms to match environmentally or imposed day and night cycle. The molecular oscillator in both the master and peripheral clocks is thought to rely on a transcriptional negative feedback loop. At the molecular level, the circadian rhythm is composed of a set of interlocked clock proteins in which CLOCK (Circadian Locomotor Output Cycles Kaput) and BMAL1 (brain and muscle ARNT-like 1) are the core positive circadian proteins. CLOCK and BMAL1 can associate with each other to form a heterodimer, which then bind to E-box in the promoter region of the target genes to induce the transcription of period (Per1, 2 and 3) and cryptochrome (Cry1 and 2) in the positive limb (5-7) (Figure 1). Once the repressor proteins CRY and PER have reached a critical concentration, they associate with each other and is phosphorylated by CK1ε, which then they can attenuate the activity of CLOCK-BMAL1 heterodimers and thereby inhibit the transcription of their own genes (8,9). To start a new cycle, the repression of CLOCK-BMAL1 heterodimer has to be released by the proteolytic degradation of PER and CRY (10,11). In addition, CLOCK-BMAL1 heterodimers induce a second regulatory loop activating transcription of retinoic acid-related orphan nuclear receptors, Rer-erbα and Rorα. Both of these nuclear receptors regulate the expression of Bmal1 through binding to the Bmal1 promoter at REV-ERBα and RORα response elements (ROREs). REV-ERBα repress the expression of Bmal1, while RORα activate the transcriptional progress (12,13). In the third regulatory loop, DEC1 (Differentiated embryo-chondrocyte expressed gene 1, also known as SHARP-2 or Stra13) and DEC2 (also known as Sharp1) are induced by the CLOCK-BMAL1 complex, subsequently DEC1 and DEC2 repress CLOCK-BMAL1-mediated transcription of Per in mice, forming a weaker negative feedback loop compared to that of PER-CRY (14). Another core member of the mammalian circadian clock in positive feedback loop is neuronal PAS-domain protein 2 (NPAS2), which is a paralogue of the CLOCK protein. NPAS2 can heterodimerize with BMAL1 to bind with E-box motifs and transcriptionally activate circadian genes, such as Per1, Per2 and Cry1 (15). NPAS2 shares similar activities with CLOCK but differs in tissue distribution. CLOCK works as a circadian protein in almost all living organisms, while NPAS2 functions as a molecular clock in mammary forebrain (15). Further regulation is achieved by post-translational modification and chromatin remodeling, which are pivotal to maintaining circadian rhythms.
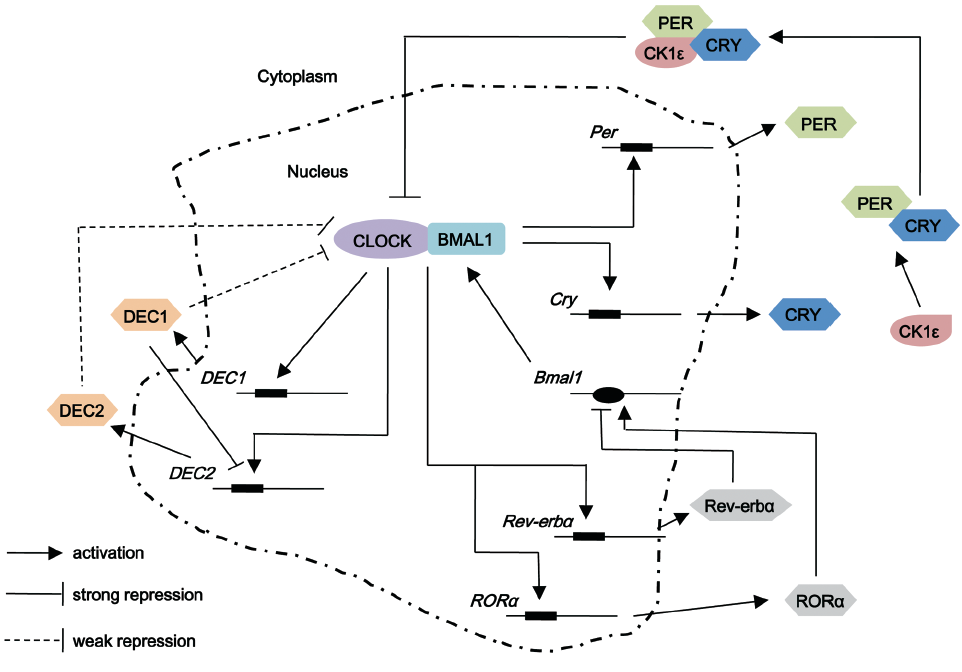
 , stands for E-box element which is present in promoter regions of the genes under consideration to which CLOCK-BMAL1 heterodimer binds;
, stands for E-box element which is present in promoter regions of the genes under consideration to which CLOCK-BMAL1 heterodimer binds;  , stands for ROREs which are retinoic acid-related orphan nuclear receptor response elements located in Bmal1 promoter for REV-ERB and ROR to bind. Casein kinase (CK) isoforms phosphorylate several circadian proteins, such as PER, CRY and BMAL1 reducing their stability and maintain the cycling function of circadian rhythm
, stands for ROREs which are retinoic acid-related orphan nuclear receptor response elements located in Bmal1 promoter for REV-ERB and ROR to bind. Casein kinase (CK) isoforms phosphorylate several circadian proteins, such as PER, CRY and BMAL1 reducing their stability and maintain the cycling function of circadian rhythmEffects of members of positive feedback loop in breast cancer
The relationship between CLOCK and estrogen signaling pathway
Clinical data shows that breast tissue from a healthy body has significantly lower CLOCK gene expression than breast tissue from patients diagnosed with breast cancer, including adjacent normal tissue, as well as invasive carcinoma and cystic change (16). These data suggest that the aberrant over-expression of the CLOCK protein may be an early event in cancer development. In addition, patients with ER-negative tumors have higher levels of CLOCK gene expression than patients with ER-positive tumors (16). Estrogen receptor (ER)-positive breast cancers generally have a better prognosis and are often responsive to anti-estrogen therapy, which is the first example of a successful therapy that targets a nuclear receptor (17). Unfortunately ER-negative breast cancers are more aggressive and are unresponsive to anti-estrogens (18). It appears that the expression of CLOCK is positively correlated with the malignancy of breast cancer. Our previous finding reveals that CLOCK promoted the proliferation of MCF-7 and T47D cell which are all ER-positive breast cancer cells by activating the transcriptional activity of ERα in the presence of estrogen (19). This may suggest that stimulation of ERα by CLOCK may represent an important mechanism for the growth of ER-positive breast cancer. As for the higher level of CLOCK observed in ERα negative tumors found in previous research (16), there may exist another unknown pathway participated by CLOCK, which has yet to be determined.
Cell-cycle genes are regulated by CLOCK-BMAL1 complex
CLOCK can not only participate in ER-related signaling pathway to regulate the proliferation of MCF-7 cells, but also activate the expression of various cell-cycle genes which contain the E-box that is the binding site for CLOCK-BMAL1. It is worth noting that a number of cell-cycle genes involved in either G2-M or G1-S transition contain E boxes in their promoter regions. It has been shown that CLOCK-BMAL1 directly regulates cell-cycle genes involved in either G2-M or G0-G1 transition. Wee-1 is one of the cell-cycle genes working as a G2-M transition checkpoint, which is directly regulated by CLOCK-BMAL1 (20,21). Wee-1 encodes a cell-cycle protein kinase that phosphorylates the CDC2/Cyclin B1 complex, causing its inactivation and delay of mitosis or arrest of the cell cycle at the G2-M interface when ongoing DNA replication contains DNA damage (21). Strikingly, elevated levels of Wee-1 in Cry mutant mice, which lack inhibition of CLOCK-BMAL1 by CRY, causes phosphorylation of CDC2/CYCB1 complex at an increased rate even in non-stressed cells, thus slowing down the G2-M transition and the overall growth rate (21,22). In addition to regulating the expression of Wee1, CLOCK/BMAL1 also regulates other cell cycle genes, including c-myc. The expression of c-myc plays a crucial role in both cell proliferation and apoptosis while the dyregulation of this gene causes genomic instability, uncontrolled cell proliferation and immortalization (23). Normally, the binding of CLOCK-BMAL1 to the E-box of c-Myc promoter represses the transcription of the c-Myc gene. However in Per2 mutants, since PER2 promotes the transcription of Bmal1 gene in addition to repressing the activity of CLOCK-BMAL1, the reduced level of BMAL1 causes upregulation of c-Myc expression(10,24). Thus this over-expression of c-Myc leads to genomic DNA damage and as a result causes hyperplasia and tumorigenesis (25).
The possible effect of HAT activity of CLOCK in the regulation of cell functions
CLOCK is considered to be an enzyme with intrinsic histone acetylase (HAT) activity, which can acetylate histones leading to chromatin remodeling or non-histone targets such as its heterodimer BMAL1, which play crucial roles in maintaining circadian rhythm (26,27). Several cell proliferation proteins, including p53 and c-Myc, and transcription factors such as ERα, NF-κB and c-Jun are acetylated by HATs (28-32). All these data show that HATs can influence cell proliferation, apoptosis, and gene transcription in multi-signal pathways different from its roles in chromatin remodeling. In addition to our previous finding that CLOCK can associate with ERα, CLOCK also works as a positive regulator of NF-κB transcription by mutual interaction (33). Thus, CLOCK can cause a significant effect in the regulation of cell functions by direct association with regulation proteins, resulting in acetylation of these proteins and possibly in influencing their activities.
The repressive role of NPAS2 in breast cancer
NPAS2, a core circadian gene and transcription regulator, is significantly associated with decreased risk of breast cancer. Mutagen (methyl methanesulfonate, MMS)-treated MCF-7 cells with normal NPAS2 expression are more likely to be in G1 or G2 phase, corresponding to the major cell cycle checkpoints for DNA damage repair, while the number of cells in S phase is significantly reduced. However, among cells with reduced NPAS2 expression by siRNA, there is no significant difference between cells treated with or without MMS, suggesting an aberrant response to DNA damage (34). Moreover, the reduced expression of NPAS2 significantly represses the expression of several cell cycle and DNA repair genes such as MAPK12 and EXO1 (34). In addition, NPAS2-BMAL1 complex has been considered to mediate the binding to the promoter of the oncogene c-myc and inhibit its transcription (25). Thus, NPAS2 may play a role in tumorigenesis by affecting expression of oncogenes and could be considered a novel putative tumor suppressor. In addition, some previous genetic epidemiological studies have shown that significant associations between a missense polymorphism (Ala394Thr) in NPAS2 and an increased risk of cancer development such as breast cancer and prostate cancer (35,36).
Therefore, although CLOCK and NPAS2 play similar roles in regulation of circadian rhythm, they cause different effects in the generation and development of breast cancer. Hypermethylation in CLOCK promoter reduce the risk of breast cancer showing that upregulation of CLOCK may promote the generation of breast cancer, while downregulation of NPAS2 decrease the sensitivity of cells to mutagen induction cell cycle arrest (34). The decrease in sensitivity contributes to cells exceeding the cell cycle checkpoint and entering into mitosis stage with DNA damage, thus leading to tumorigenesis of the breast cells.
Implication of PERs family in breast cancer
Clinical studies have shown that the expression of all three Per genes is deregulated in breast cancer patients and 95% of breast tumors display nearly absent or dyregulated levels of PER1 and PER2 in tumor cells compared with adjacent normal cells (37). Moreover, a structural variation of the Per3 gene has been identified as a potential biomarker for breast cancer in pre-menopausal women (1). More importantly, PER1 and PER2 appear to act as tumor suppressors in mice (25,38). Therefore, all three PER proteins are proposed to function as tumor suppressors.
PER1 and repair of DNA damage
PER1 can work as a tumor suppressor by regulating cell cycle genes and associating with key DNA damage activated checkpoint proteins. Over-expression of Per1 in cancer cells increases ionizing radiation-induced apoptosis (38). ATM kinase and its downstream effector Chk2 are activated by DNA double-strand break (DBS) and in turn phosphorylate a network of proteins that initiate DNA repair, cell cycle arrest and apoptosis. In addition to regulating cell cycle genes, PER1 directly associate with DBS-activated kinases CHK2 and ATM and this interaction is necessary for efficient activation of Chk2 in response to DBS (38). Collectively, PER1 play a key role in the DNA damage response by interacting with and activating the ATM checkpoint pathway. Thus, PER1 can work as a tumor suppressor through multiple signaling activations.
Relationship between PER2 and cell cycle
The transcript pattern of c-myc, which plays crucial roles in both cell proliferation and apoptosis, shows a low-amplitude circadian oscillation in all mouse tissues; however, the expression is significantly increased throughout the 24-hour period in Per2 mutants (39). In contrast, the transcription of p53, which plays a critical role in the G1-S checkpoint, is decreased in Per2 mutants. Following γ radiation, cells over-expressed with c-myc are less efficient in G1 arrest compared with normal cells, revealing that over-expression of c-MYC could contribute cells to overcome cell cycle in the presence of genomic DNA damage (39,40). On the contrary, the loss of Per2 partially impairs p53-mediated apoptosis, resulting in accumulation of damaged cells following γ radiation. A mutation (S662G) in PER2 is responsible for familial advanced sleep phase syndrome (41), while recent data shows that this mutation site (S662G) is also linked to cell cycle progression and tumorigenesis. PER2 (S662G) mutation leads to enhanced resistance to X-ray-induced apoptosis and increased E1A and RAS related oncogenic transformation (42). At the same time, over-expressed PER2 in mice mammary carcinoma cell line (EMT6) led to reduced cellular proliferation and rapid apoptosis, but not in non-tumorigenic NIH3T3 cells (43). The temporal expression of genes involved in cell cycle regulation and tumor suppression, including c-Myc, cyclin A, Mdm-2, and Gadd45α, is deregulated in Per2 mutant mice (44). Down-regulation of PER2 increases the cell cycle protein level of cyclin D1 and cyclin E and promoted the growth of cultured mice mammary tumor cells as well as tumors in mice (45).
PER2 participates in estrogen signaling directly
Besides CLOCK, which directly participates in the breast cancer related estrogen-signaling pathway, PER2 is another circadian rhythm regulating protein. Per2 expression is now known to be induced by estrogen, and PER2 can bind to ERα and enhance its degradation, resulting in significant growth inhibition, loss of clonogenic ability and cellular apoptosis (46). PER2 can be induced by E2, and it in turn is shown to promote the expression of BMAL1 by forming regulatory complexes with RORα and Rev-erbα via binding with ROREs in BMAL1 promoter, forming a PER2-BMAL1 loop (47). An abnormal PER2-BMAL1 loop can impair estrogen-regulated morphogenetic processes due to lack of proper circadian availability of ERα in HME1 cells (3). All these data links the circadian rhythm to ERα signaling pathway.
The roles of DEC family in the proliferation and metastasis of breast cancer cells
DEC1 and DEC2 are basic helix-loop-helix transcriptional factors, reportedly involved in maintaining circadian rhythm of cells, cellular growth, cellular differentiation, cellular apoptosis and cellular tumorigenesis (48). The expression of DEC1 is considered to be correlated with increased malignant potential and invasiveness of tumor cells. The expression of DEC1 increases on progression from normal to in situ and invasive breast carcinoma, showing a significant positive correlation between DEC1 and tumor grade. DEC1 expression is strongly induced by hypoxia, the hypoxically induced protein angiogenin and tumor grade, revealing there is an effect of the level of DEC1 in suppression of tumor differentiation and potential apoptosis (49,50). The reduced level of DEC1 results in the increasing expression of claudin-1 which functions as a tumor invasion suppressor in MCF-7 and MDA-MB-231 breast cancer cell line. Thus, DEC1 may promote the invasive ability of breast cancer cells by the downregulation of claudin-1 (51). DEC1 itself can be induced by TGF-β, which facilitates tumor growth and metastasis in advanced cancer stages and represses apoptosis of mice mammary carcinoma cells (52). Additionally, a dominant-negative mutant of DEC1 (dnDEC1) prevents lung and liver metastasis of breast cancer cells in vivo (52). However, other researchers show that DEC1 is a SUMOylated protein, and its SUMOylation enhances its ability to inhibit the transcription level of cyclin D1, which promotes the proliferation of breast cancer cells and mediate cell cycle arrest in G1 phase in fibroblast cells (53,54). In conclusion, there is a wide range of consensus on the role of overexpressed DEC1 in promoting the metastasis ability of breast cancer cells, whereas there is still the ongoing debate on the effect of DEC1 on the proliferation of cells. It is possible that DEC1 have different functions in different environments. DEC1 is abundantly expressed in tumors and protects these diseased cells against induced apoptosis with serum starvation by upregulating the expression of antiapoptotic protein surviving (55). Since tumor cells, especially malignant tumors can usually survive in hypoxia and low nutrition environment. Overexpressed DEC1 may repress cell proliferation in normal conditions, but in a hostile environment, such as hypoxia or low nutrition, DEC1 starts a distinct anti-pressure pathway to inhibit cell apoptosis via upregulating antiapoptotic proteins.
Overexpression of cyclin D1 induces mammary tumorigenesis, and higher levels of cyclin D1 in ER-positive breast cancer patients are associated with poorer prognoses. Unlike DEC1, overexpression of DEC2 dramatically inhibits cell proliferation and represses the expression of cyclin D1 in human mammary epithelial cells (56). In addition to playing roles in cell proliferation, DEC2 also takes effect in breast cancer metastasis. DEC2 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors-1α (HIF-1α). DEC2 is a crucial regulator of the invasive and metastatic phenotype in triple-negative breast cancer (TNBC), which is one of the most aggressive types of breast cancer. The cell migration in vitro, invasive, or metastatic behaviours is dependent on HIF-1α protein. Thus, DEC2 suppresses TNBC metastasis by associating with HIF-1α and promoting the HIF proteasomal degradation (57).
Therefore, although DEC1 and DEC2 are members of the same family, they have different effects on the regulation of cell functions. Depending on the situation, DEC1 plays distinct roles in cell proliferation. As for the effect on cell metastasis, DEC1 promotes its progression, while DEC2 represses this progress. Previous studies show that DEC1 was expressed markedly higher in carcinomas, whereas DEC2 was expressed markedly higher in the adjacent normal tissues (58). At the same time, DEC1 negatively regulates the expression of DEC2 by direct DNA binding to the E-box in the proximal promoter of DEC2 (58). Thus, we speculate that homeostatic levels of these two circadian proteins are disrupted in some breast cancers, especially in high grade invasive tumors.
Chronotherapy
Chronotherapy against cancers are dependent on the effect that the circadian rhythm exerts on multiple cellular processes, such as cell cycle, DNA repair, proliferation and apoptosis, and drug metabolism, which are crucial molecular determinants of cellular pharmacokinetics and pharmacodynamics of cytotoxia/cytostatic drugs (59). Chronotherapy is a treatment strategy that searches for the optimal time for drug administration in accordance with the body biological clock, in order to promote the therapeutic effect of anti-cancer drugs.
Research has shown that the growth rate of breast cancer is regulated by the circadian clock with two daily peaks, coupled to the daily expression patterns of clock-controlled genes that regulate cell proliferation, while down-expression of PER1 promote growth of tumors via enhancing the amplitude of the two daily growth peaks (60). These data shows that PER1 represses tumor growth only at specific times of a day.
In malignant tumors, vascular endothelial growth factor (VEGF) is expressed at substantially increased levels, and its expression is often related with poor prognosis in several types of tumors. The transcription of VEGF gene is controlled by the circadian molecular clockwork in hypoxic tumor cells and showing a circadian oscillation in the cancer cells implanted in mice. It is notable that the circadian variation of VEGF influences the pharmacological efficacy of antiangiogenic agents, since angiogenesis inhibitors exhibit the most repressive effect on the growth of tumors when the drug is administered at the expression peak of VEGF (61). Damaging the circadian rhythm also influences response to anticancer drugs in mice. The sensitivity of wild-type mice varies largely depending on the time of anticancer drug cyclophosphamide (CY) treatment, while Clock mutant and Bmal1 knockout mice show a high sensitivity to administration at all times tested. Contrastingly, mice with Cry1-/-Cry2-/- double knockouts exhibit more resistance to CY compared with wild type mice (62). This resistance reveals that response to anticancer drug CY is directly related with functional module of the major circadian clock.
A recent study suggests a molecular relationship between circadian rhythm and oral drug absorption. The expression of BCRP (breast cancer resistance protein) limits systemic exposure to xenobiotics. Alterations in the function and expression of this protein could account for part of the variation in oral drug absorption and is regulated under control of circadian clock-ATF4 pathway. This data shows that the circadian clock-ATF4 pathway causes the oscillation of BCRP function and induces the circadian change in intestinal drug absorption (63). Anticancer drugs generally produce their cytotoxic effect in both normal and malignant tissues. If we could identify the difference of circadian rhythm of intestine absorption capacity between normal and cancer tissues, we can prognosticate the most favorable time for drug administration, which is when intestine absorption ability is low in normal tissues, and when the ability is high in cancer tissues.
Conclusions
Over past few decades, hormone therapy has been the most effective treatment for women with ER-positive breast cancer (64,65). However, the appearance of resistance to hormone therapy has promoted the search for a different strategy, including focusing on the relationship between the disruption of circadian rhythm and breast tumorgenesis. Circadian regulation is pivotal to maintaining normal cellular physiology, and the disruption of circadian rhythm is closely related with multiple diseases, such as diabetes, obesity, dyssomnias, and cancer. Most living organisms exhibit behavioural and physiological circadian oscillations. According to previous research, when compared to those of normal breast cells, these oscillations are usually damaged in breast cancer cells. Exploring the difference in the circadian rhythm between normal tissues and malignant tissues not only contribute to investigating the mechanism behind the generation and development of cancers, but also this disparity in the circadian rhythm between normal and tumor cells is crucial to maximizing the effect of anticancer drugs and supports the academic foundation for tumor chronotherapy.
Acknowledgments
This work was supported by grants (31171353, 31271500 to HW) from National Natural Science Foundation of China and grants (973 program: 2011CB504201 to HW) from the Ministry of Science and Technology of China.
Disclosure: The authors declare no conflict of interest.
References
- Zhu Y, Brown HN, Zhang Y, et al. Period3 structural variation: a circadian biomarker associated with breast cancer in young women. Cancer Epidemiol Biomarkers Prev 2005;14:268-70. [PubMed]
- Viswanathan AN, Schernhammer ES. Circulating melatonin and the risk of breast and endometrial cancer in women. Cancer Lett 2009;281:1-7. [PubMed]
- Rossetti S, Corlazzoli F, Gregorski A, et al. Identification of an estrogen-regulated circadian mechanism necessary for breast acinar morphogenesis. Cell Cycle 2012;11:3691-700. [PubMed]
- Rossetti S, Esposito J, Corlazzoli F, et al. Entrainment of breast (cancer) epithelial cells detects distinct circadian oscillation patterns for clock and hormone receptor genes. Cell Cycle 2012;11:350-60. [PubMed]
- Dunlap JC. Molecular bases for circadian clocks. Cell 1999;96:271-90. [PubMed]
- Lowrey PL, Takahashi JS. Genetics of the mammalian circadian system: Photic entrainment, circadian pacemaker mechanisms, and posttranslational regulation. Annu Rev Genet 2000;34:533-62. [PubMed]
- Young MW, Kay SA. Time zones: a comparative genetics of circadian clocks. Nat Rev Genet 2001;2:702-15. [PubMed]
- Takano A, Isojima Y, Nagai K. Identification of mPer1 phosphorylation sites responsible for the nuclear entry. J Biol Chem 2004;279:32578-85. [PubMed]
- Eide EJ, Woolf MF, Kang H, et al. Control of mammalian circadian rhythm by CKIepsilon-regulated proteasome-mediated PER2 degradation. Mol Cell Biol 2005;25:2795-807. [PubMed]
- Shearman LP, Sriram S, Weaver DR, et al. Interacting molecular loops in the mammalian circadian clock. Science 2000;288:1013-9. [PubMed]
- Vitaterna MH, Selby CP, Todo T, et al. Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc Natl Acad Sci U S A 1999;96:12114-9. [PubMed]
- Akashi M, Takumi T. The orphan nuclear receptor RORalpha regulates circadian transcription of the mammalian core-clock Bmal1. Nat Struct Mol Biol 2005;12:441-8. [PubMed]
- Preitner N, Damiola F, Lopez-Molina L, et al. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 2002;110:251-60. [PubMed]
- Honma S, Kawamoto T, Takagi Y, et al. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature 2002;419:841-4. [PubMed]
- Reick M, Garcia JA, Dudley C, et al. NPAS2: an analog of clock operative in the mammalian forebrain. Science 2001;293:506-9. [PubMed]
- Hoffman AE, Yi CH, Zheng T, et al. CLOCK in breast tumorigenesis: genetic, epigenetic, and transcriptional profiling analyses. Cancer Res 2010;70:1459-68. [PubMed]
- Osborne CK. Steroid hormone receptors in breast cancer management. Breast Cancer Res Treat 1998;51:227-38. [PubMed]
- Sheikh MS, Garcia M, Pujol P, et al. Why are estrogen-receptor-negative breast cancers more aggressive than the estrogen-receptor-positive breast cancers? Invasion Metastasis 1994-1995;14:329-36. [PubMed]
- Li S, Wang M, Ao X, et al. CLOCK is a substrate of SUMO and sumoylation of CLOCK upregulates the transcriptional activity of estrogen receptor-α. Oncogene 2012. [Epub ahead of print]. [PubMed]
- Hirayama J, Cardone L, Doi M, et al. Common pathways in circadian and cell cycle clocks: light-dependent activation of Fos/AP-1 in zebrafish controls CRY-1a and WEE-1. Proc Natl Acad Sci U S A 2005;102:10194-9. [PubMed]
- Matsuo T, Yamaguchi S, Mitsui S, et al. Control mechanism of the circadian clock for timing of cell division in vivo. Science 2003;302:255-9. [PubMed]
- Oishi K, Miyazaki K, Kadota K, et al. Genome-wide expression analysis of mouse liver reveals CLOCK-regulated circadian output genes. J Biol Chem 2003;278:41519-27. [PubMed]
- Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol 2006;16:318-30. [PubMed]
- Hogenesch JB, Panda S, Kay S, et al. Circadian transcriptional output in the SCN and liver of the mouse. Novartis Found Symp 2003;253:171-80; discussion 52-5, 102-9, 180-3 passim.
- Fu L, Pelicano H, Liu J, et al. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 2002;111:41-50. [PubMed]
- Hirayama J, Sahar S, Grimaldi B, et al. CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature 2007;450:1086-90. [PubMed]
- Doi M, Hirayama J, Sassone-Corsi P. Circadian regulator CLOCK is a histone acetyltransferase. Cell 2006;125:497-508. [PubMed]
- Kim MY, Woo EM, Chong YT, et al. Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol Endocrinol 2006;20:1479-93. [PubMed]
- Hoberg JE, Popko AE, Ramsey CS, et al. IkappaB kinase alpha-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol Cell Biol 2006;26:457-71. [PubMed]
- Raivich G. c-Jun expression, activation and function in neural cell death, inflammation and repair. J Neurochem 2008;107:898-906. [PubMed]
- Mao B, Zhao G, Lv X, et al. Sirt1 deacetylates c-Myc and promotes c-Myc/Max association. Int J Biochem Cell Biol 2011;43:1573-81. [PubMed]
- Brochier C, Dennis G, Rivieccio MA, et al. Specific acetylation of p53 by HDAC inhibition prevents DNA damage-induced apoptosis in neurons. J Neurosci 2013;33:8621-32. [PubMed]
- Spengler ML, Kuropatwinski KK, Comas M, et al. Core circadian protein CLOCK is a positive regulator of NF-κB-mediated transcription. Proc Natl Acad Sci U S A 2012;109:E2457-65. [PubMed]
- Hoffman AE, Zheng T, Ba Y, et al. The circadian gene NPAS2, a putative tumor suppressor, is involved in DNA damage response. Mol Cancer Res 2008;6:1461-8. [PubMed]
- Zhu Y, Stevens RG, Leaderer D, et al. Non-synonymous polymorphisms in the circadian gene NPAS2 and breast cancer risk. Breast Cancer Res Treat 2008;107:421-5. [PubMed]
- Chu LW, Zhu Y, Yu K, et al. Variants in circadian genes and prostate cancer risk: a population-based study in China. Prostate Cancer Prostatic Dis 2008;11:342-8. [PubMed]
- Chen ST, Choo KB, Hou MF, et al. Deregulated expression of the PER1, PER2 and PER3 genes in breast cancers. Carcinogenesis 2005;26:1241-6. [PubMed]
- Gery S, Komatsu N, Baldjyan L, et al. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol Cell 2006;22:375-82. [PubMed]
- Sheen JH, Dickson RB. Overexpression of c-Myc alters G(1)/S arrest following ionizing radiation. Mol Cell Biol 2002;22:1819-33. [PubMed]
- Vafa O, Wade M, Kern S, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell 2002;9:1031-44. [PubMed]
- Xu Y, Toh KL, Jones CR, et al. Modeling of a human circadian mutation yields insights into clock regulation by PER2. Cell 2007;128:59-70. [PubMed]
- Gu X, Xing L, Shi G, et al. The circadian mutation PER2(S662G) is linked to cell cycle progression and tumorigenesis. Cell Death Differ 2012;19:397-405. [PubMed]
- Hua H, Wang Y, Wan C, et al. Circadian gene mPer2 overexpression induces cancer cell apoptosis. Cancer Sci 2006;97:589-96. [PubMed]
- Lee CC. Tumor suppression by the mammalian Period genes. Cancer Causes Control 2006;17:525-30. [PubMed]
- Yang X, Wood PA, Oh EY, et al. Down regulation of circadian clock gene Period 2 accelerates breast cancer growth by altering its daily growth rhythm. Breast Cancer Res Treat 2009;117:423-31. [PubMed]
- Gery S, Virk RK, Chumakov K, et al. The clock gene Per2 links the circadian system to the estrogen receptor. Oncogene 2007;26:7916-20. [PubMed]
- Schmutz I, Ripperger JA, Baeriswyl-Aebischer S, et al. The mammalian clock component PERIOD2 coordinates circadian output by interaction with nuclear receptors. Genes Dev 2010;24:345-57. [PubMed]
- Liu Y, Sato F, Kawamoto T, et al. Anti-apoptotic effect of the basic helix-loop-helix (bHLH) transcription factor DEC2 in human breast cancer cells. Genes Cells 2010;15:315-25. [PubMed]
- Wykoff CC, Pugh CW, Maxwell PH, et al. Identification of novel hypoxia dependent and independent target genes of the von Hippel-Lindau (VHL) tumour suppressor by mRNA differential expression profiling. Oncogene 2000;19:6297-305. [PubMed]
- Chakrabarti J, Turley H, Campo L, et al. The transcription factor DEC1 (stra13, SHARP2) is associated with the hypoxic response and high tumour grade in human breast cancers. Br J Cancer 2004;91:954-8. [PubMed]
- Liu Y, Miao Y, Wang J, et al. DEC1 is positively associated with the malignant phenotype of invasive breast cancers and negatively correlated with the expression of claudin-1. Int J Mol Med 2013;31:855-60. [PubMed]
- Ehata S, Hanyu A, Hayashi M, et al. Transforming growth factor-beta promotes survival of mammary carcinoma cells through induction of antiapoptotic transcription factor DEC1. Cancer Res 2007;67:9694-703. [PubMed]
- Wang Y, Rao VK, Kok WK, et al. SUMO modification of Stra13 is required for repression of cyclin D1 expression and cellular growth arrest. PLoS One 2012;7:e43137. [PubMed]
- Hong Y, Xing X, Li S, et al. SUMOylation of DEC1 protein regulates its transcriptional activity and enhances its stability. PLoS One 2011;6:e23046. [PubMed]
- Li Y, Xie M, Yang J, et al. The expression of antiapoptotic protein survivin is transcriptionally upregulated by DEC1 primarily through multiple sp1 binding sites in the proximal promoter. Oncogene 2006;25:3296-306. [PubMed]
- Li Y, Shen Q, Kim HT, et al. The rexinoid bexarotene represses cyclin D1 transcription by inducing the DEC2 transcriptional repressor. Breast Cancer Res Treat 2011;128:667-77. [PubMed]
- Montagner M, Enzo E, Forcato M, et al. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nature 2012;487:380-4. [PubMed]
- Li Y, Xie M, Song X, et al. DEC1 negatively regulates the expression of DEC2 through binding to the E-box in the proximal promoter. J Biol Chem 2003;278:16899-907. [PubMed]
- Innominato PF, Lévi FA, Bjarnason GA. Chronotherapy and the molecular clock: Clinical implications in oncology. Adv Drug Deliv Rev 2010;62:979-1001. [PubMed]
- Yang X, Wood PA, Ansell CM, et al. The circadian clock gene Per1 suppresses cancer cell proliferation and tumor growth at specific times of day. Chronobiol Int 2009;26:1323-39. [PubMed]
- Koyanagi S, Kuramoto Y, Nakagawa H, et al. A molecular mechanism regulating circadian expression of vascular endothelial growth factor in tumor cells. Cancer Res 2003;63:7277-83. [PubMed]
- Gorbacheva VY, Kondratov RV, Zhang R, et al. Circadian sensitivity to the chemotherapeutic agent cyclophosphamide depends on the functional status of the CLOCK/BMAL1 transactivation complex. Proc Natl Acad Sci U S A 2005;102:3407-12. [PubMed]
- Hamdan AM, Koyanagi S, Wada E, et al. Intestinal expression of mouse Abcg2/breast cancer resistance protein (BCRP) gene is under control of circadian clock-activating transcription factor-4 pathway. J Biol Chem 2012;287:17224-31. [PubMed]
- Li S, Yang C, Hong Y, et al. The transcriptional activity of co-activator AIB1 is regulated by the SUMO E3 ligase PIAS1. Biol Cell 2012;104:287-96. [PubMed]
- Chang AK, Wu H. The role of AIB1 in breast cancer. Oncol Lett 2012;4:588-94. [PubMed]