
A systemic review of glutathione S-transferase P1 Ile105Val polymorphism and colorectal cancer risk
Introduction
Colorectal cancer (CRC) is one of the commonest human malignant diseases and the leading causes of cancer-related death in western countries, accounting for approximately 9% of all cancer incidence and mortality (1). Although early diagnosis and timely operation may benefit patients and result in a relative complete healing, about 25% of newly diagnosed patients were metastatic CRC, for whose 5-year survival is 11% (2). Thus, the reliable genetic tests are already used to detect high-penetrance alleles of genes, such as APC and DNA mismatch repair genes, in order to screen CRC high-risk groups. Study based on the analysis of phenotype in twins has improved that genetic factors were attributed to about 35% of CRC development (3), however, the high-penetrance genes only account for 5% of all CRCs (4). Epidermiological studies have demonstrated that numerous low-penetrance alleles contributing to CRC risk. There is also evidence that susceptibility to CRC is mediated by alterations in the detoxifying enzyme system (5), since CRC is a complicated disease which is determined by multiple exposures of endogeneous and dietary carcinogens. The glutathione S-transferases (GST) supergene family of phase II metabolic enzymes, play an important role in detoxifying carcinogens in cellular defense system. Glutathione S-transferase P1 (GSTP1), which is expressed in normal colon epithelial tissue and overexpressed in tumor colon and rectum (6,7), plays a major role in GST family. Polymorphism of a transversion of adenine to guanine substitution at base pair 313 which leads to substitution of isoleucine (Ile) with valine (Val) at condon 105 has been improved to affect activity of GSTP1 (8). It is supposed that individuals with GSTP1 of low enzymatic activity could be associated with increased risk of CRC. However, the relation between GSTP1 Ile105Val and CRC susceptibility is still controversial (9-12). The difficulty of searching the relation between GSTP1 Ile105Val and CRC susceptibility could be due to the modest effect of single nucleotide polymorphism (SNP), small sample studies are lack of power and fail to verify the association. Nevertheless, meta-analysis, which is a statistical method to combine data together for more powerful estimation of true effect, could clarify inconclusive results in genetic association studies. Yong Gao and colleagues have evaluated the predictive GSTP1 Ile105Val and CRC risk but fail to provide a clear conclusion (13). In the last few years, a number of high-quality large-sample studies were conducted to investigate the relevance of GSTP1 Ile105Val with CRC risk. So we conducted a new meta-analysis, combining results from previously published articles to draw a more precise conclusion of the relation between GSTP1 Ile105Val and CRC susceptibility.
Materials and methods
To ensure the precise of our meta-analysis, we reported it on the basis of the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) statement (shown in Table S1) (http://www.prisma-statement.org).

Full table
Publication search
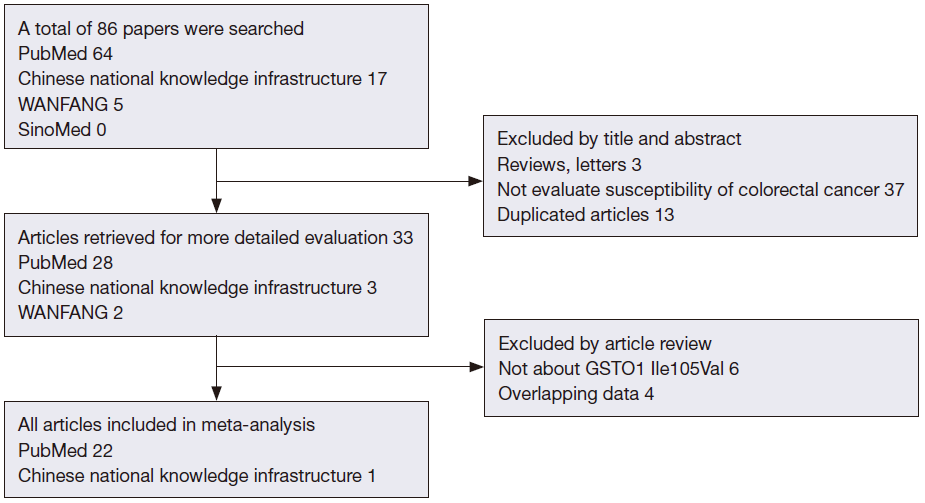
Systematic computerized searches of the PubMed, Chinese National Knowledge Infrastructure, WANFANG and SinoMed (up to July 4, 2013) were performed. Following search terms were utilized: “colorectal neoplasms”, “polymorphism, single nucleotide”, “Genetic Predisposition to Disease”, “Glutathione S-Transferase pi” and “rs1695”. The search was limited to human studies. All eligible studies were retrieved, and their bibliographies were checked for other relevant publications.
Inclusion and exclusion criteria
The included studies have to meet the following criteria: (I) the case-control study focused on the relationship between GSTP1 Ile105Val and risk of CRC; (II) providing adequate data for pooled analyses, including total number of CRC cases and controls, as same as the number of cases and controls for each genotypes; (III) studies with full text articles. Exclusion criteria included: (I) reviews, tutorials and letters; (II) not case-control studies; (III) animal studies; (IV) insufficient data were reported as number of cases and controls without genotype data; (V) duplicate data. When the same patient population was used in several publications, only the most recent, largest or complete study was included in the meta-analysis.
Data extraction
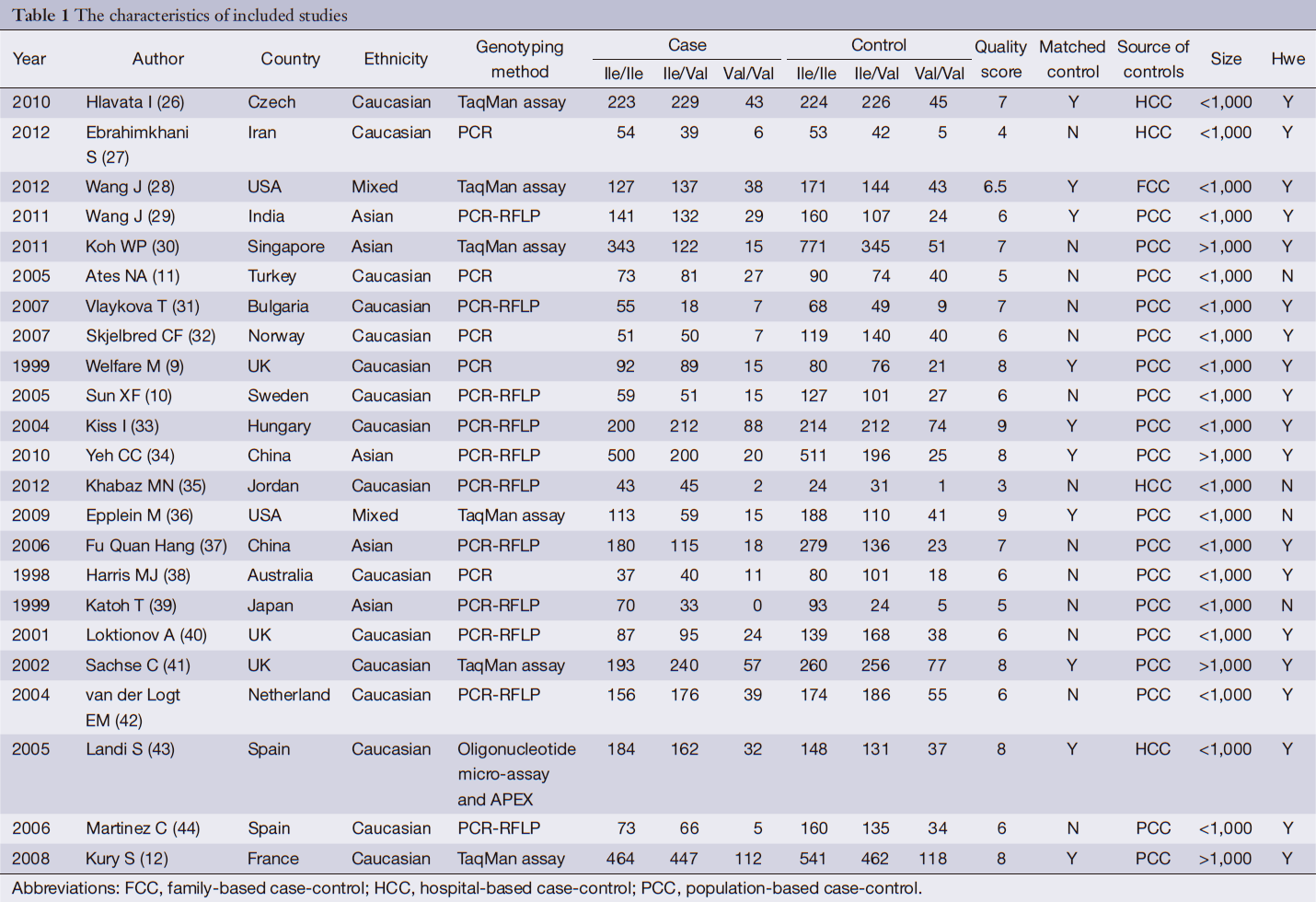
Information was carefully extracted from all eligible studies. The following data were collected from each study: first author’s name, year of publication, country, ethnicity of participants, sources of controls [family-based case-control (FCC), hospital-based case-control (HCC) or population-based case-control (PCC)], number of cases and controls, genotyping methods, genotypes, goodness-in-fitness of Hardy-Weinberg equilibrium (HWE) and matched control. HCC study was defined as controls from hospitalization patient, PCC was from healthy people, and FCC was from patients’ family. Matched control study was defines as controls matched by at least three variables: age, gender and region. Data extraction was done independently by three of the authors (Qi-Bin Song, Wei-Guo Hu). Disagreement was resolved by discussion between the three authors.
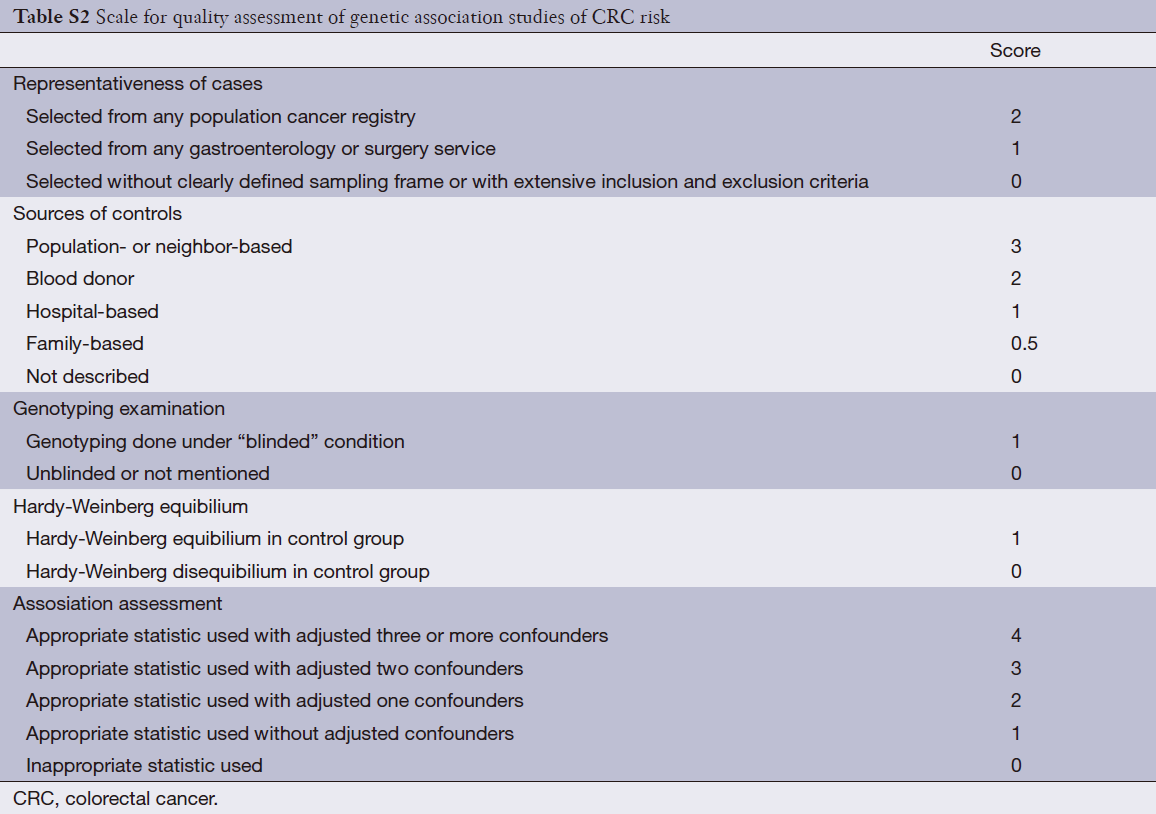
Quality score assessment
The quality of articles was independently assessed by the same three reviewers (Qi-Bin Song, Wei-Guo Hu). The quality score assessment was adopted from predefined criteria established by meta-analysis of molecular association studies (14-16). The criteria included representativeness of cases, sources of controls, genotyping examination, HWE and association assessment (see in Table S2). Scores ranged from the lowest zero to highest eleven. Studies with the score more than 5 were suggested as “moderate or high quality” ones, while those lower than 5 (include 5) were considered as “low quality” ones.
Full table
Statistical methods
We assessed HWE in the controls for each study using chi-square test at first, and P value <0.05 was considered as a significant disequilibrium (17). Odds ratio (OR) and corresponding 95% confidence interval (95% CI) were employed to assess the strength of associations between GSTP1 Ile105Val and risk of CRC. The wild type Ile/Ile was considered as a reference. The genetic comparisons included homozygous model (Val/Val vs. Ile/Ile), heterozygous model (Ile/Val vs. Ile/Ile), dominant model (Val/Val + Ile/Val vs. Ile/Ile) and recessive model (Val/Val vs. Ile/Val + Ile/Ile). We also conducted subgroup analyses by ethnicity, source of control (HCC, PCC or FCC), sample size (<1,000 or >1,000) and matched control (Yes/No).
Heterogeneity was checked by a chi-square-based Q-test (18,19) and I2 statistic (18). Heterogeneity was considered significantly when the P value of Q-test was less than 0.1. The following thresholds were used for I2 statistic: I2 =0-25%, no heterogeneity; I2 =25-50%, moderate heterogeneity; I2 =50-75%, large heterogeneity; I2 =75-100%, extreme heterogeneity. If significant heterogeneity was found (P<0.10 or I2 >50%), the random-effects model (the DerSimonian and Laird method) (20) instead of the fixed-effects model (the Mantel-Haenszel method) (21) was used for further analysis. A Galbraith plot was used to assess the extent of heterogeneity between studies from meta-analyses (22). To investigate the possible sources of the heterogeneity, we performed meta-regression analyses based on following aspects: published year, ethnicity, source of control (HCC, PCC or FCC), study sample size (<1,000 or >1,000), HWE in control (Yes/No) and matched control (Yes/No). For purpose of examining the influence of single study on the pooled OR and assessing stability of the results, sensitivity analysis was performed to repeat analyses by omitting one study at a time.
Funnel plots were used to explore the presence of publication bias. The degree of funnel plot asymmetry was assessed by Begg’s (23) and Egger’s test (24). The trim-and-fill method was implemented to evaluate number of potentially missing studies and assess the effect of publication bias on meta-analysis (25). All P values are two-tailed with a significant level at 0.05. All the statistical tests used in our meta-analysis were performed with STATA version 10.0 (Stata Corporation, College Station, TX, USA).
Results
Characteristics of the included studies
After exclusion of duplicate and irrelevant studies (Figure 1), 23 studies including 6,981 cases and 8,977 controls comparing the GSTP1 Ile105Val and susceptibility of CRC were identified according to the inclusion criteria in the meta-analysis. Four studies showed mixed ethnicity. Four articles were based on large sample size (>1,000). In these studies, 18 were PCC, 4 were HCC and 1 was FCC. The detailed information of these articles was listed in Table 1.
Full table
Overall meta-analysis and further subgroup analysis
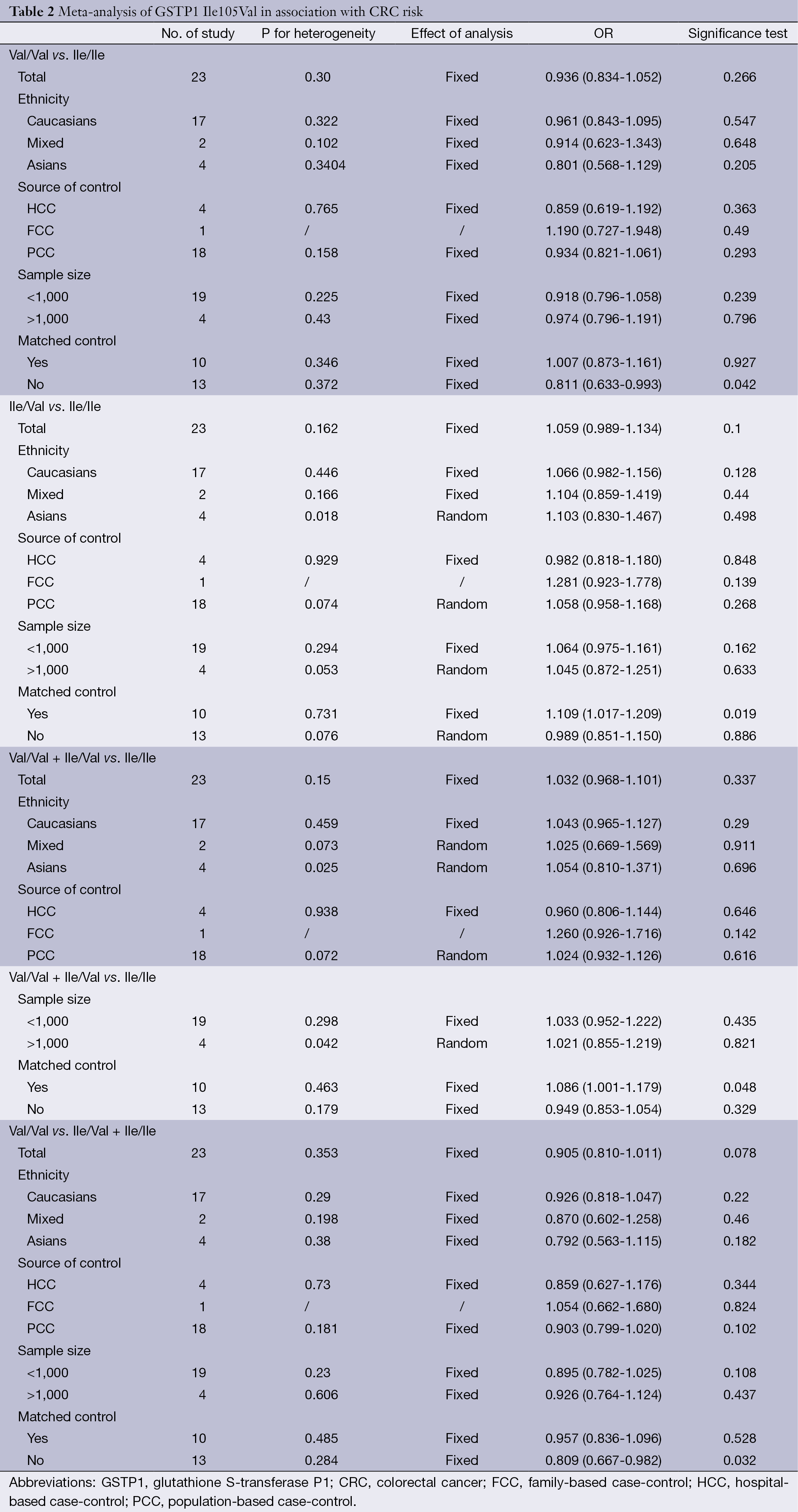
A total of 23 case-control studies with 6,981 cases and 8,977 controls were included in the analyses. Table 2 listed the main results of the pooled analysis. Overall, the results of meta-analyses suggested that GSTP1 Ile105Val was not related to risk of CRC (Val/Val vs. Ile/Ile, OR =0.94, 95% CI =0.83-1.05; Ile/Val vs. Ile/Ile, OR =1.06, 95% CI =0.99-1.13; Val/Val + Ile/Val vs. Ile/Ile dominant model, OR =1.03, 95% CI =0.97-1.10; Val/Val vs. Ile/Val + Ile/Ile recessive model, OR =0.91, 95% CI =0.81-1.01), without between-study heterogeneity. In subgroup analyses, the variant allele Val was associated with susceptibility of CRC in matched articles (Ile/Val vs. Ile/Ile: OR =1.11, 95% CI =1.02-1.21; Val/Val + Ile/Val vs. Ile/Ile: OR =1.09, 95% CI =1.00-1.18). It was confused that Val/Val was correlated with decreased CRC risk of non-matched articles in heterozygous model and recessive model. This might be in correlated with bias caused by low-quality studies. So we removed the low-quality studies (quality score ≤5) and conducted new meta-analyses.
Full table
The specific results were listed in Table 3. As shown in Table 3, no associations were observed in the homozygous model (OR =0.945, 95% CI =0.839-1.065), as well as in other three models (heterozygous model: OR =1.050, 95% CI =0.979-1.127; dominant model: OR =1.027, 95% CI =0.961-1.099; recessive model: OR =0.918, 95% CI =0.819-1.029). Subgroup analyses were conducted according to ethnicity, source of control, sample size and matched control. Different ethnicities were classified as Caucasians, Asians and mixed races. There were no statistically significant findings among Caucasians, Asians and mixed races in all genetic comparisons. In the subgroup analyses based on source of control, sample size and matched control, the variant allele Val was not related with susceptibility of CRC in all subgroups except in matched control studies. In the studies with matched controls, the variant allele Val had significantly relationship with increased risk of CRC (heterozygous model: OR =1.109, 95% CI =1.017-1.209, Figure 2A; dominant model: OR =1.086, 95% CI =1.001-1.179, Figure 2B).
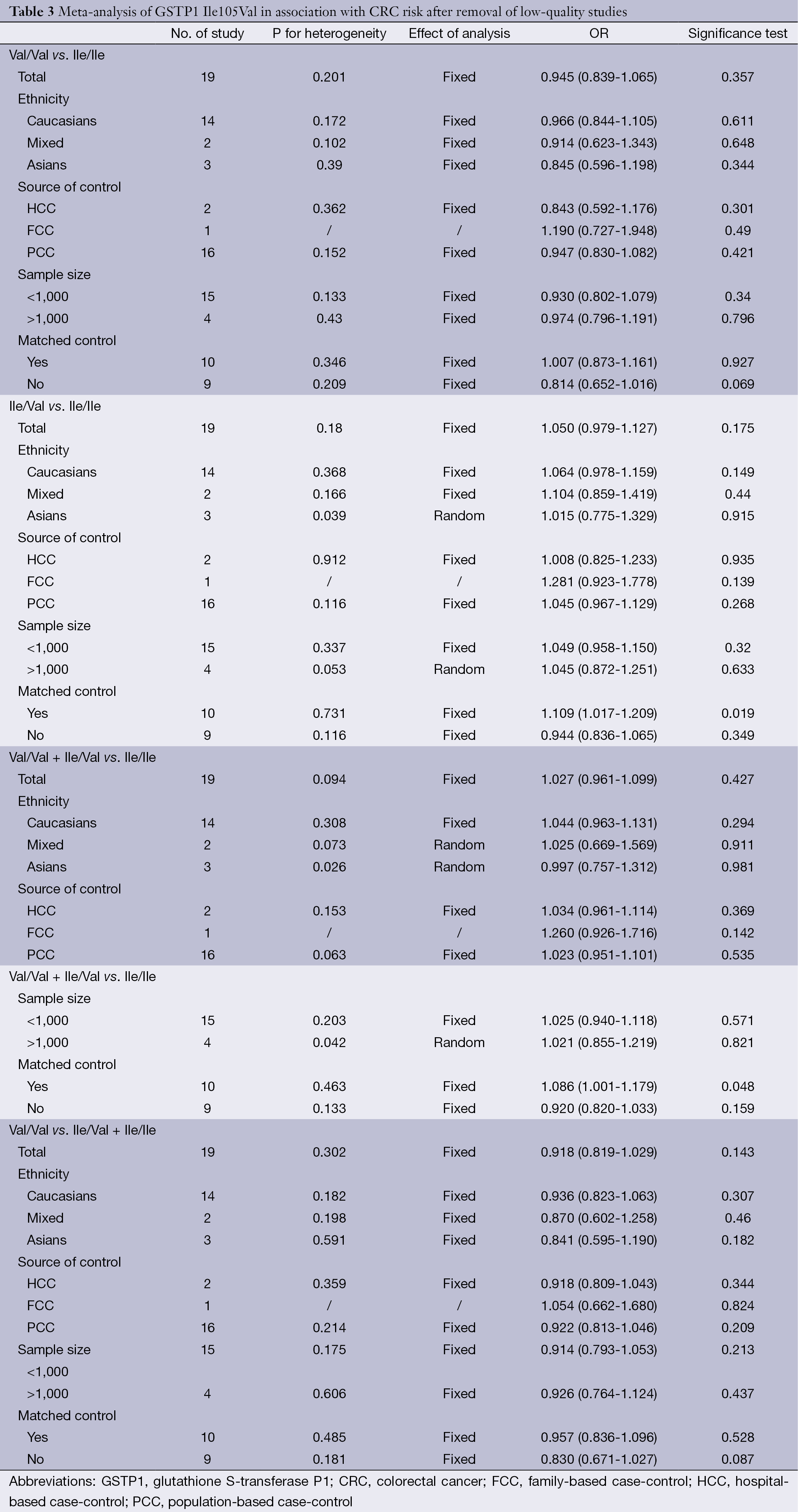
Full table
Heterogeneity analysis
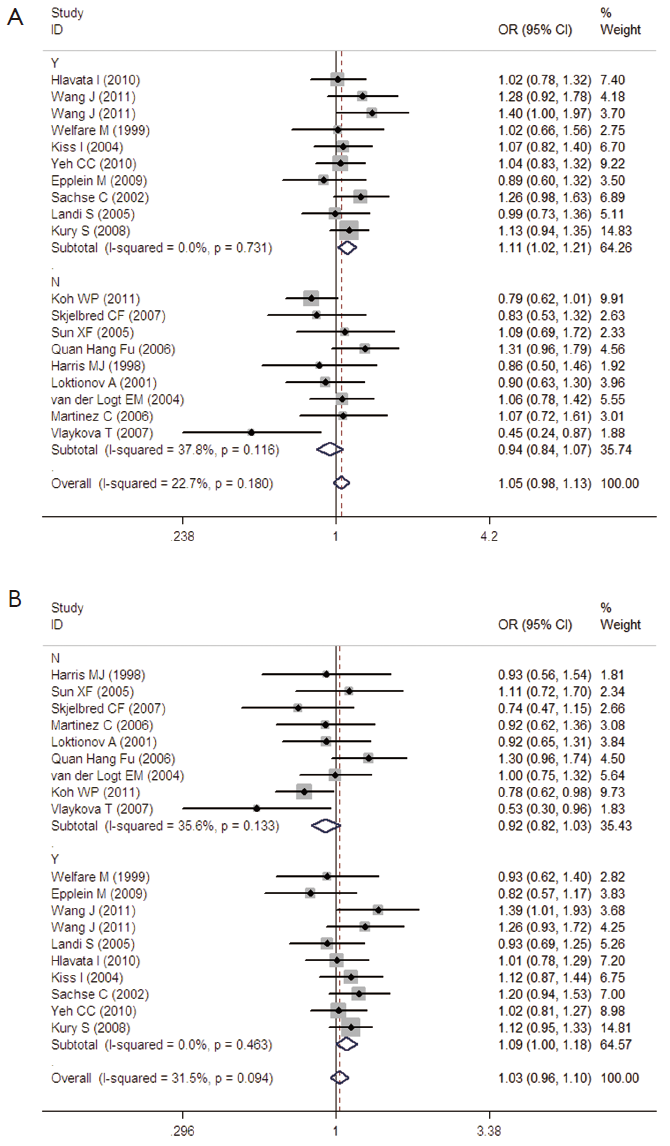
The genotype data in the 19 studies were homogenous in all genetic comparisons (homozygous model: I2 =20.8%, Pheterogeneity =0.201; heterozygous model: I2 =22.7%, Pheterogeneity =0.180; dominant model: I2 =31.5%, Pheterogeneity =0.094; recessive model: I2 =12.5%, Pheterogeneity =0.302). However, the heterogeneity remained in subgroup analyses. So we conducted Galbraith plot analyses of included studies to assess the potential sources of heterogeneity. Martinez C (44) was the contributor of heterogeneity in the homozygous model and recessive model, while Koh WP (30) and Vlaykova T (31) were the sources of heterogeneity in the heterozygous model and dominant model (see in Figure S1A and B). The meta-regression analyses were further used to explore the sources of heterogeneity across the included studies, we assessed all genetic comparisons by published year, ethnicity, source of control, sample size, HWE in control and matched control. We performed an empty meta-regression to estimate the baseline value of tau2, and the univariate model was conducted by the above aspects. In the univariate analysis, the results suggested matched controls were attributed to heterogeneity and reduced the tau2 value from 0.0043 to 0 in the heterozygous comparison and from 0.0088 to 0 .0027 in the dominant comparison.
Sensitivity analysis
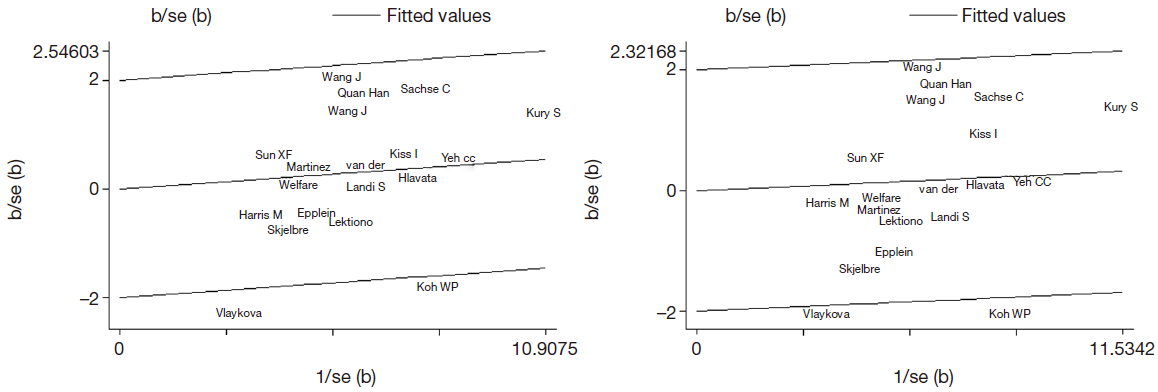
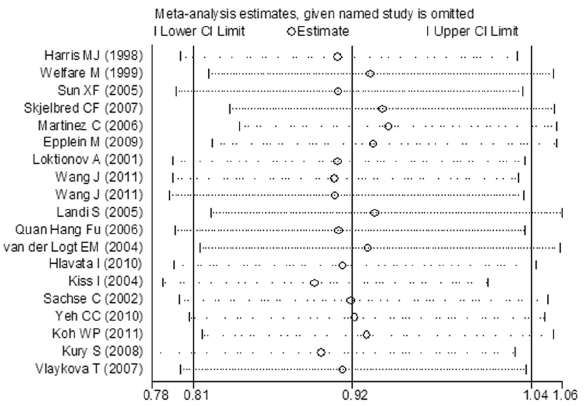
Sensitivity analyses were performed under random-effects model to examine the influence of single study on the pooled value and assess stability of the results. In the Val/Val vs. Ile/Ile model, the most influencing study seemed to be the study conducted by Kury S (12), the OR was 0.945 (95% CI =0.839-1.065) and 0.911 (95% CI =0.781-1.063) before and after removing the study. Koh WP (30) had a critical influence on the results in heterozygous comparison and dominant comparison. The OR was 1.050 (95% CI =0.979-1.127) and 1.077 (95% CI =0.998-1.162), and 1.027 (95% CI =0.961-1.099) and 1.047 (95% CI =0.968-1.133) before and after removing the study in heterozygous and dominant comparison, respectively. The most influencing study in the recessive study was conducted by Kiss I (33), the OR was 0.893 (95% CI =0.787-1.013) after removing it (see in Figure S2). Removal of a single study did not impact on the pooled results in all genetic comparisons, the sensitivity analyses supported the robustness of the current meta-analyses.
Publication bias
The funnel plot, Begg’s test and Egger’s test were used to explore the publication bias. The funnel plots were symmetrical in general in heterozygous, dominant models and recessive model (see in Figure S3A, B and C). The Begg’s test and Egger’s test showed no evidence of publication bias in meta-analyses (heterozygous model: Begg’s test P=0.124, Egger’s test P=0.135; dominant model: Begg’s test P=0.142, Egger’s test P=0.112; recessive model: Begg’s test P=0.184, Egger’s test P=0.079). However, Begg’s and Egger’s tests revealed that there might be some unpublished positive articles, especially some small sample size studies, were not included in the meta-analyses of homozygous models (Begg’s test P=0.036, Egger’s test P=0.032). Then the trim-and-fill method was used to estimate the number of missing studies resulting from publication bias. In the homozygous model, there was no trimming study was performed and no difference between random-effects and fixed-effects model, indicating the results were not greatly influenced by publication bias and our meta-analyses were statistically robust.
Discussion
CRC is usually identified as a complex multi-factor, multi-variable disease, which is determined by exposures to carcinogens and individual genetic background (45). Previous studies have revealed cigarette smoking, diets high in red meat and fat are associated with increased risk of CRC (46,47). The metabolites of cigarette and high-fat foods, polycyclic aromatic hydrocarbons (PAHs), are complex carbon molecules known as strong carcinogens which form oxidation DNA adducts, induce gene mutation and lead to cell malignant transformation (48). It is supposed that susceptibility to CRC is mediated by genes involved in detoxifying enzyme system, especially genes with PAH metabolism. GSTP1, a major member of GST family, plays an important role in CRC susceptibility. Polymorphism of a transversion of adenine to guanine substitution at base pair 313 which leads to substitution of Ile with Val at condon 105 has been improved to affect activity of GSTP1 (8). Some studies have indicated the activity of variant Val allele to metabolite carcinogens is lower than that of Ile allele (49,50). Thus, it is indicated that individuals with GSTP1 Val allele of low enzymatic activity could be in relevance with increased risk of CRC. Harris examined prediction of GSTP1 Ile105Val in CRC risk in 1998 (38), since then, numerous studies attempted to explore the relation but failed to provide precise conclusion. Chen (51) and Gao (13) carried out meta-analyses and found out no connection of GSTP1 Ile105Val to CRC risk. In the last few years, a number of high-quality large-sample studies were conducted to investigate the relevance of GSTP1 Ile105Val to CRC. Based on the cumulative evidence, we carried out an updating meta-analysis to draw a precise conclusion.
We conducted the meta-analysis including 23 case-control studies of 6,981 cases and 8,977 controls comparing the GSTP1 Ile105Val and susceptibility of CRC. When subgroup analyses were performed by ethnicity, source of control, sample size and matched control, significant association was observed between GSTP1 Ile105Val and CRC. However, it is confused that Val allele was related with decreased risk in unmatched controls under homozygous comparison and recessive comparison, but with increased risk in matched controls under heterozygous comparison and dominant comparison. This might be in correlated with bias caused by low-quality studies. So we removed the low-quality studies and conducted new meta-analyses. Further meta-analyses found GSTP1 Ile105Val polymorphism was associated with increased CRC risk in matched controls under heterozygous comparison and dominant comparison. The meta-regression analyses were further conducted to explore sources of heterogeneity. In all possible influential factors including published year, ethnicity, source of control, sample size, HWE in control and matched control, results suggested matched controls were the significant factor influencing between-study heterogeneity. Further sensitivity analyses suggested the results were persistent and robust. Publication bias was found in homozygous comparisons. We carried out trim-and-fill method to estimate the number of missing studies resulting from publication bias. There was no trimming study was performed and no difference between random-effects and fixed-effects model. Taken together, we found that GSTP1 Ile108Val polymorphism might be related with increased risk of CRC, but it still requires a lot of high-quality case-control studies to confirm.
It is thought that the high dose should exert the more significant effect in a viewpoint of dose-response relationship. Interestingly, we found that GSTP1 Ile105Val heterozygotes instead of homozygotes had a significant increased risk of CRC. The variant heterozygotes may have damaged three dimensional structures and are limited with detoxifying function. Another possible interpretation was the heterozygotes may be in linkage disequilibrium with other loci in relevance with CRC risk. The similar findings were described by Ma and Liu (52,53). Ma and colleagues found a significant increased risk of breast cancer was related with variant CDKN1B C-79T heterozygotes, but not homozygotes. Meanwhile, Liu found EPHX1 His139Arg heterozygotes, other than homozygotes, had a significant relation with CRC risk.
Despite the strength of our study that yielded enough power, that’s a lot of room for improvement. At first, CRC is a complex disease, which is resulting from interactions among environmental factors and genetic factors. However, lacking the individual personal data and environmental data limited us to explore the interaction between other possible exposures and GSTP1 Ile105Val on susceptibility of CRC. Further studies should focus on the mechanism of CRC risk, especially gene-gene and gene-environment interactions. Additionally, the quality of included studies is uneven. The relation between GSTP1 Ile105Val polymorphism and CRC risk is contradictory at first. When excluding the low-quality studies, GSTP1 Ile105Val is associated with increased risk of CRC only limited in studies with matched control. Included studies with high-quality will provide reliable data and drawn a precise conclusion.
Conclusions
In conclusion, the results from our meta-analysis provide a comprehensive description of relation between GSTP1 Ile105Val and CRC susceptibility. It is indicated that variant Val allele is associated with increased risk of CRC limited in matched control studies. However, more high-quality case-control studies should be performed to confirm the authenticity of the relation between GSTP1 Ile105Val and CRC susceptibility. Since other factors, such as environmental carcinogens and genetic background, also have impact on CRC susceptibility, gene-gene and gene-environment interactions should be carried on research in order to make clear the mechanism of CRC risk.
Acknowledgements
Disclosure: The authors declare no conflict of interest.
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013;63:11-30. [PubMed]
- Meyerhardt JA, Mayer RJ. Systemic therapy for colorectal cancer. N Engl J Med 2005;352:476-87. [PubMed]
- Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 2000;343:78-85. [PubMed]
- de la Chapelle A. Genetic predisposition to colorectal cancer. Nat Rev Cancer 2004;4:769-80. [PubMed]
- Inoue H, Kiyohara C, Shinomiya S, et al. Glutathione S-transferase polymorphisms and risk of colorectal adenomas. Cancer Lett 2001;163:201-6. [PubMed]
- Doğru-Abbasoğlu S, Mutlu-Turkoglu U, Turkoglu S, et al. Glutathione S-transferase-pi in malignant tissues and plasma of human colorectal and gastric cancers. J Cancer Res Clin Oncol 2002;128:91-5. [PubMed]
- Ranganathan S, Tew KD. Immunohistochemical localization of glutathione S-transferases alpha, mu, and pi in normal tissue and carcinomas from human colon. Carcinogenesis 1991;12:2383-7. [PubMed]
- Zimniak P, Nanduri B, Pikula S, et al. Naturally occurring human glutathione S-transferase GSTP1-1 isoforms with isoleucine and valine in position 104 differ in enzymic properties. Eur J Biochem 1994;224:893-9. [PubMed]
- Welfare M, Monesola AA, Bassendine MF, et al. Polymorphisms in GSTP1, GSTM1, and GSTT1 and susceptibility to colorectal cancer. Cancer Epidemiol Biomarkers Prev 1999;8:289-92. [PubMed]
- Sun XF, Ahmadi A, Arbman G, et al. Polymorphisms in sulfotransferase 1A1 and glutathione S-transferase P1 genes in relation to colorectal cancer risk and patients’ survival. World J Gastroenterol 2005;11:6875-9. [PubMed]
- Ateş NA, Tamer L, Ates C, et al. Glutathione S-transferase M1, T1, P1 genotypes and risk for development of colorectal cancer. Biochem Genet 2005;43:149-63. [PubMed]
- Küry S, Buecher B, Robiou-Du-Pont S, et al. Low-penetrance alleles predisposing to sporadic colorectal cancers: a French case-controlled genetic association study. BMC Cancer 2008;8:326. [PubMed]
- Gao Y, Pan X, Su T, et al. Glutathione S-transferase P1 Ile105Val polymorphism and colorectal cancer risk: a meta-analysis and HuGE review. Eur J Cancer 2009;45:3303-14. [PubMed]
- Thakkinstian A, Mcevoy M, Minelli C, et al. Systematic review and meta-analysis of the association between {beta}2-adrenoceptor polymorphisms and asthma: a HuGE review. Am J Epidemiol 2005;162:201-11. [PubMed]
- Camargo MC, Mera R, Correa P, et al. Interleukin-1beta and interleukin-1 receptor antagonist gene polymorphisms and gastric cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2006;15:1674-87. [PubMed]
- Thakkinstian A, D’Este C, Eisman J, et al. Meta-analysis of molecular association studies: vitamin D receptor gene polymorphisms and BMD as a case study. J Bone Miner Res 2004;19:419-28. [PubMed]
- Haber M. Exact significance levels of goodness-of-fit tests for the Hardy-Weinberg equilibrium. Hum Hered 1981;31:161-6. [PubMed]
- Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med 2002;21:1539-58. [PubMed]
- Higgins JP, Thompson SG, Deeks JJ, et al. Measuring inconsistency in meta-analyses. BMJ 2003;327:557-60. [PubMed]
- DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials 1986;7:177-88. [PubMed]
- Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 1959;22:719-48. [PubMed]
- Galbraith RF. A note on graphical presentation of estimated odds ratios from several clinical trials. Stat Med 1988;7:889-94. [PubMed]
- Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics 1994;50:1088-101. [PubMed]
- Egger M, Davey SG, Schneider M, et al. Bias in meta-analysis detected by a simple, graphical test. BMJ 1997;315:629-34. [PubMed]
- Song F, Gilbody S. Bias in meta-analysis detected by a simple, graphical test. Increase in studies of publication bias coincided with increasing use of meta-analysis. BMJ 1998;316:471. [PubMed]
- Hlavata I, Vrana D, Smerhovsky Z, et al. Association between exposure-relevant polymorphisms in CYP1B1, EPHX1, NQO1, GSTM1, GSTP1 and GSTT1 and risk of colorectal cancer in a Czech population. Oncol Rep 2010;24:1347-53. [PubMed]
- Ebrahimkhani S, Asgharian AM, Nourinaier B, et al. Association of GSTM1, GSTT1, GSTP1 and CYP2E1 single nucleotide polymorphisms with colorectal cancer in Iran. Pathol Oncol Res 2012;18:651-6. [PubMed]
- Wang J, Joshi AD, Corral R, et al. Carcinogen metabolism genes, red meat and poultry intake, and colorectal cancer risk. Int J Cancer 2012;130:1898-907. [PubMed]
- Wang J, Jiang J, Zhao Y, et al. Genetic polymorphisms of glutathione S-transferase genes and susceptibility to colorectal cancer: a case-control study in an Indian population. Cancer Epidemiol 2011;35:66-72. [PubMed]
- Koh WP, Nelson HH, Yuan JM, et al. Glutathione S-transferase (GST) gene polymorphisms, cigarette smoking and colorectal cancer risk among Chinese in Singapore. Carcinogenesis 2011;32:1507-11. [PubMed]
- Vlaykova T, Miteva L, Gulubova M, et al. Ile105Val GSTP1 polymorphism and susceptibility to colorectal carcinoma in Bulgarian population. Int J Colorectal Dis 2007;22:1209-15. [PubMed]
- Skjelbred CF, Saebo M, Hjartaker A, et al. Meat, vegetables and genetic polymorphisms and the risk of colorectal carcinomas and adenomas. BMC Cancer 2007;7:228. [PubMed]
- Kiss I, Nemeth A, Bogner B, et al. Polymorphisms of glutathione-S-transferase and arylamine N-acetyltransferase enzymes and susceptibility to colorectal cancer. Anticancer Res 2004;24:3965-70. [PubMed]
- Yeh CC, Lai CY, Hsieh LL, et al. Protein carbonyl levels, glutathione S-transferase polymorphisms and risk of colorectal cancer. Carcinogenesis 2010;31:228-33. [PubMed]
- Khabaz MN. The GSTP1 Ile105Val polymorphism is not associated with susceptibility to colorectal cancer. Asian Pac J Cancer Prev 2012;13:2949-53. [PubMed]
- Epplein M, Wilkens LR, Tiirikainen M, et al. Urinary isothiocyanates; glutathione S-transferase M1, T1, and P1 polymorphisms; and risk of colorectal cancer: the Multiethnic Cohort Study. Cancer Epidemiol Biomarkers Prev 2009;18:314-20. [PubMed]
- Fu QH, Gao CM, Wu JZ, et al. Polymorphism of GSTT1, GSTM1 and GSTP1 and Susceptibility of Colorectal Cancer. Pract J Cancer 2006;247-50.
- Harris MJ, Coggan M, Langton L, et al. Polymorphism of the Pi class glutathione S-transferase in normal populations and cancer patients. Pharmacogenetics 1998;8:27-31. [PubMed]
- Katoh T, Kaneko S, Takasawa S, et al. Human glutathione S-transferase P1 polymorphism and susceptibility to smoking related epithelial cancer; oral, lung, gastric, colorectal and urothelial cancer. Pharmacogenetics 1999;9:165-9. [PubMed]
- Loktionov A, Watson MA, Gunter M, et al. Glutathione-S-transferase gene polymorphisms in colorectal cancer patients: interaction between GSTM1 and GSTM3 allele variants as a risk-modulating factor. Carcinogenesis 2001;22:1053-60. [PubMed]
- Sachse C, Smith G, Wilkie MJ, et al. A pharmacogenetic study to investigate the role of dietary carcinogens in the etiology of colorectal cancer. Carcinogenesis 2002;23:1839-49. [PubMed]
- van der Logt EM, Bergevoet SM, Roelofs HM, et al. Genetic polymorphisms in UDP-glucuronosyltransferases and glutathione S-transferases and colorectal cancer risk. Carcinogenesis 2004;25:2407-15. [PubMed]
- Landi S, Gemignani F, Moreno V, et al. A comprehensive analysis of phase I and phase II metabolism gene polymorphisms and risk of colorectal cancer. Pharmacogenet Genomics 2005;15:535-46. [PubMed]
- Martínez C, Martin F, Fernandez JM, et al. Glutathione S-transferases mu 1, theta 1, pi 1, alpha 1 and mu 3 genetic polymorphisms and the risk of colorectal and gastric cancers in humans. Pharmacogenomics 2006;7:711-8. [PubMed]
- Ishibe N, Sinha R, Hein DW, et al. Genetic polymorphisms in heterocyclic amine metabolism and risk of colorectal adenomas. Pharmacogenetics 2002;12:145-50. [PubMed]
- Gertig DM, Hunter DJ. Genes and environment in the etiology of colorectal cancer. Semin Cancer Biol 1998;8:285-98. [PubMed]
- Greenwald P, Clifford CK, Milner JA. Diet and cancer prevention. Eur J Cancer 2001;37:948-65. [PubMed]
- Perera FP, Mooney LA, Dickey CP, et al. Molecular epidemiology in environmental carcinogenesis. Environ Health Perspect 1996;104 Suppl 3:441-3. [PubMed]
- Sundberg K, Johansson AS, Stenberg G, et al. Differences in the catalytic efficiencies of allelic variants of glutathione transferase P1-1 towards carcinogenic diol epoxides of polycyclic aromatic hydrocarbons. Carcinogenesis 1998;19:433-6. [PubMed]
- Johansson AS, Stenberg G, Widersten M, et al. Structure-activity relationships and thermal stability of human glutathione transferase P1-1 governed by the H-site residue 105. J Mol Biol 1998;278:687-98. [PubMed]
- Chen K, Jiang QT, He HQ. Relationship between metabolic enzyme polymorphism and colorectal cancer. World J Gastroenterol 2005;11:331-5. [PubMed]
- Ma H, Jin G, Hu Z, et al. Variant genotypes of CDKN1A and CDKN1B are associated with an increased risk of breast cancer in Chinese women. Int J Cancer 2006;119:2173-8. [PubMed]
- Liu F, Yuan D, Wei Y, et al. Systematic review and meta-analysis of the relationship between EPHX1 polymorphisms and colorectal cancer risk. PLoS One 2012;7:e43821. [PubMed]