
Is new American Thyroid Association risk classification for hereditary medullary thyroid carcinoma applicable to Chinese patients? A single-center study
Introduction
Hereditary medullary thyroid carcinoma (MTC), one of the main clinical manifestations of multiple endocrine neoplasia type 2 (MEN2), is autosomal dominant. Other common symptoms of MEN2 include pheochromocytoma (PHEO) and hyperparathyroidism (HPT). Germline mutations of rearranged during transfection (RET) proto-oncogene were found to be responsible for the development of MEN2 syndromes. Age at initial MTC diagnosis, life expectancy and overall prognosis after thyroidectomy are associated with different RET gene mutations; indeed, the RET mutation genotype determines the phenotype.
RET gene mutations are classified into 3 categories, including highest (ATA-HST), high (ATA-H) and moderate (ATA-MOD) risk groups, in the new 2015 American thyroid association (ATA) guidelines (1,2). Prophylactic surgical intervention and follow-up intervals should be tailored to the various mutation risk categories.
RET gene mutations in hereditary MTC have been rarely assessed in the Chinese population, for the correlation between genotype and phenotype. In the present study, we assessed whether the new ATA risk classification is suitable to Chinese patients with hereditary MTC. In addition, we reported our initial experience of prophylactic thyroidectomy in RET-gene mutation carriers.
Materials and methods
Patient enrollment
Patients treated in Cancer Hospital, Chinese Academy of Medical Science, from 2010 to 2016, with high probability of hereditary MTC were screened. High probability was considered when the patient: 1) had a family history of MTC and/or PHEO; 2) had no apparent familial history of endocrine neoplasia, but was initially treated at an age below 35 years; and 3) had multiple lesions or other endocrine neoplasia. This study was approved by the Ethics Committee of Cancer Hospital, Chinese Academy of Medical Science, and written informed consent was obtained from all the participants.
RET mutation detection
RET mutations in exons 8, 10, 11, 13, 14, 15 and 16 were detected in genomic DNA specimens from peripheral blood leucocytes of highly suspected hereditary MTC patients, by polymerase chain reaction (PCR) and direct DNA sequencing (3). The sequencing data were compared with the web based ARUP online Scientific Resource RET database to determine RET-gene mutation carriers. Then, the carrier’s relatives were also recruited for germline RET gene assessment. A total of 73 subjects from 22 families were recruited in this study, including 20 patients diagnosed with MTC but without gene detection for patient refusal or death. The patients with no RET mutation genotype were evaluated by taking into consideration their relatives’ data.
Follow-up and prophylactic thyroidectomy
For patients with confirmed MTC, original laboratory results, radiographic imaging data, and operative and pathology reports were reviewed. Then, they were followed up and treated as necessary. For the carriers who had never undergone surgery for thyroid disease, transcutaneous cervical ultrasonography, computed tomography (CT), and plasma basal calcitonin and carcinoembryonic antigen (CEA) level assessment were prescribed. For young carriers who were not diagnosed with MTC, prophylactic thyroidectomy was recommended according to the new ATA guidelines, and performed after consent of their parents. Prophylactic thyroidectomies were performed by an experienced surgeon (Mr. Zhang) using 2.5× magnifying glasses.
Statistical analysis
The disease stage at the initial diagnosis was used to measure the biological aggressiveness of MTC. Staging was based on the American Joint Committee on Cancer TNM Classification 7th edition. Patients with advanced disease (stage III, IV) were considered to have more aggressive disease than those with small, localized MTC (stage I or II). Univariable and multivariable logistic regression analyses were used to derive the associations of risk factors with the original TNM classification. The statistical analysis was performed with SPSS software (Version 14.0; SPSS Inc., Chicago, IL, USA).
Results
RET gene mutation and ATA risk classification
A total of 73 patients from 22 families were recruited in this study, including 53 who had undergone RET exon sequencing. Among the remaining 20 patients, 16 were diagnosed pathologically with MTC, 1 patient was diagnosed with MTC clinically but declined treatment, and 2 with a history of cervical mass died of PHEO related cardiovascular accident. An infant who died of Hirschsprung’s disease was also assessed.
According to the new ATA guidelines, the present study population included 1 patient (1 family) in the ATA-HST group, 48 (12 families) in the ATA-H group, and 24 (9 families) in the ATA-MOD group. The distribution of the identified RET mutations is shown in Table 1.
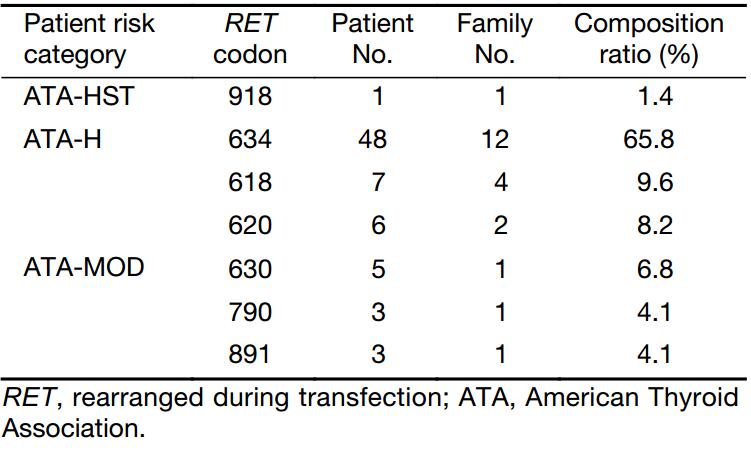
Full table
RET gene mutations and aggressiveness of hereditary MTC
General information of ATA-H & ATA-MOD groups
Except for one MEN2B patient who carried an ATA-HST mutation, the remaining 72 patients, in the ATA-H and ATA-MOD groups, had MEN2A. The clinical characteristics of MEN2A patients are shown in Table 2. In the ATA-MOD group, according to the new guidelines, 7 families were subtyped as classical MEN2A, 1 as MEN2A with Hirschsprung’s disease, and 10 as familial medullary thyroid carcinoma (FMTC). Among the patients, 1 patient died of Hirschsprung’s disease, while all the remaining 23 survived at the time of last follow-up. A total of 16 patients had undergone thyroidectomy, and the median age at initial treatment was 36.5 (range, 7.0–56.0) years. Lymph node metastases at the initial treatment were found in 8 (50.0%) of the 16 patients. At the time of diagnosis, 8 patients (50.0%) had stage 0, I, or II disease and 8 (50.0%) had stage III or IV disease. A total of 7 patients underwent a second operation because of local recurrence. At the last follow-up, 2 patients had persistent disease.
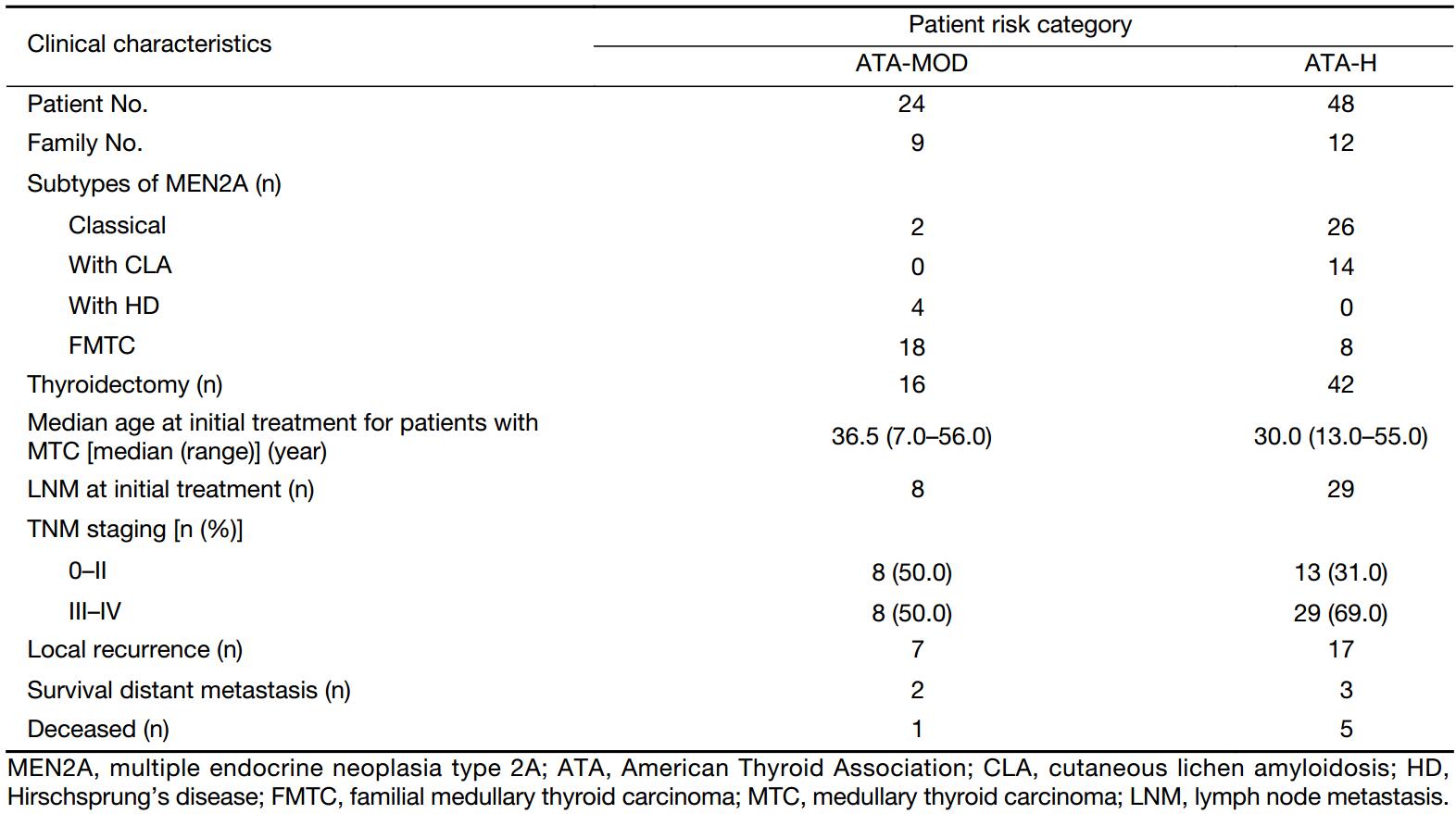
Full table
In the ATA-H group, 7 families were subtyped as classical MEN2A, 3 as MEN2A with cutaneous lichen amyloidosis (CLA), and 2 as FMTC. In this study population, 3 patients died of advanced MTC and 2 of PHEO related cardiovascular accident; the remaining 43 patients survived at the time of last follow-up. A total of 42 patients had undergone thyroidectomy, and the median age at initial treatment was 30.0 (range, 13.0–55.0) years. Lymph node metastases at the initial treatment were found in 29 (69.0%) of the 42 patients. At the time of diagnosis, 13 patients (31.0%) had stage 0, I, or II disease and 29 patients (69.0%) had stage III or IV disease. A total of 17 patients underwent a second operation due to local recurrence. At the last follow-up, 3 patients had persistent disease.
Comparison of initial treatment data between ATA-H & ATA-MOD groups
Univariate logistic regression was used to assess the associations of various risk factors and risk categories (ATA-MOD & ATA-H) with MTC aggressiveness (stage) at initial treatment. The detailed data are listed in Table 3. Age at initial treatment and risk category showed significant associations with MTC stage at initial diagnosis. The probability of advanced MTC was increased by 9.1% per year of age at initial treatment. The patients with high risk had 4.167 times higher probability than those with the modest risk of having III/IV stage disease. Gender and index case had no significant associations with disease stage at initial treatment.
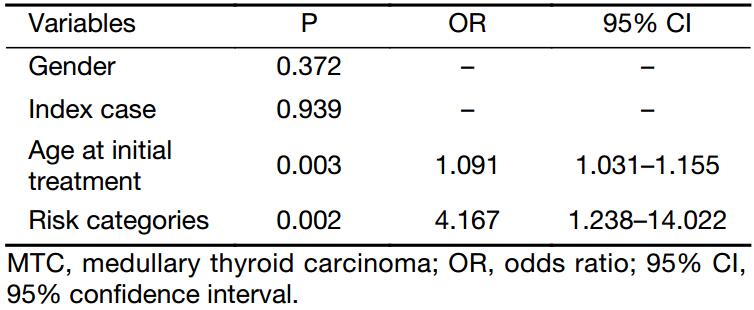
Full table
Two significant independent variables in univariate analysis were used in multivariate logistic regression analysis further, and the statistical difference was even more significant (Table 4). On the basis of multivariate analysis, the risk of having stage III or IV MTC at the time of diagnosis increased 11.6% per year of age at initial treatment. The likelihood of having stage III or IV MTC at the time of diagnosis for ATA-H group was 7.888 times higher than that of ATA-MOD group.
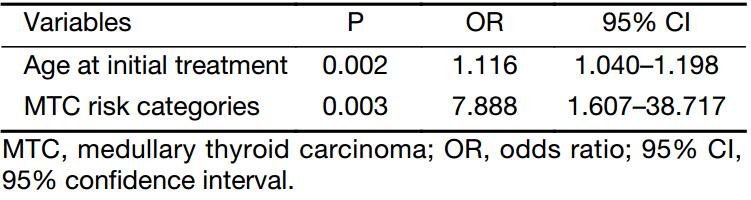
Full table
Prophylactic thyroidectomies
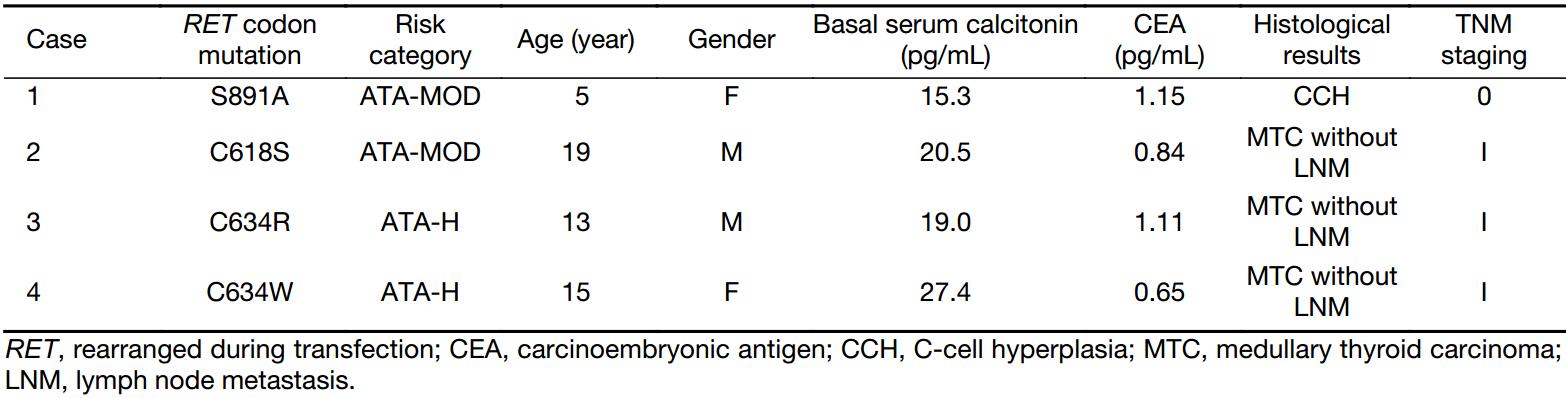
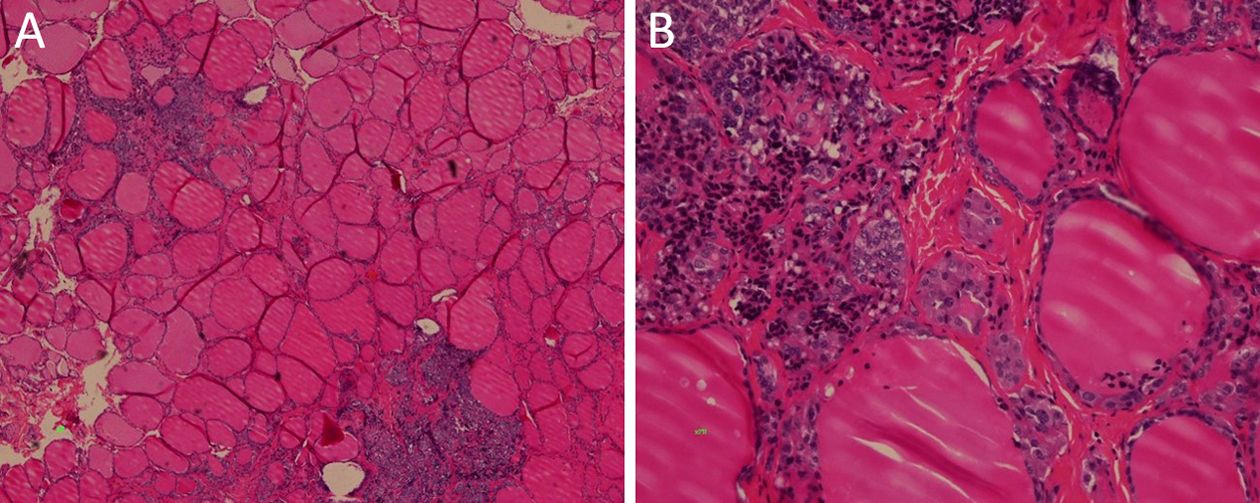
A total of 4 RET-gene mutation carriers with no apparent MTC underwent prophylactic thyroidectomy. Their clinical characteristics are shown in Table 5. Their RET codon mutations were S891A, C618S, C634R and C634W, respectively. Cases 1 & 2 were categorized as ATA-MOD; the other 2 carrying mutated codon 634 were categorized as ATA-H. Preoperative basal serum calcitonin levels were 15.3–27.4 pg/mL, which were slightly above normal levels. Prophylactic total thyroidectomy and central compartment neck dissection were performed for these patients. Except for case 1, whose histological exam revealed C-cell hyperplasia (CCH), multifocal micro MTCs (<1.0 cm) were found in thyroid specimens from cases 2–4, with no lymph node metastasis (LNM). Figure 1 shows micrographs for case 4. Multifocal CCH was detected at low magnification and MTC at high magnification. Basal serum calcitonin amounts were detected the day after surgery, and dropped to <2.0 pg/mL rapidly. The 4 patients were followed up for 6 months to 4 years. Basal serum calcitonin levels remained below 2.0 pg/mL. Cervical ultrasound found no local recurrence or metastasis.
Full table
Serum parathyroid hormone (PTH) levels were detected the day after surgery; in case of levels below the normal value (15 pg/mL), detection was performed repetitively at every follow-up. Transient hypoparathyroidism was found in cases 1 and 4. The lowest PTH levels were 2.76 pg/mL in case 1; 11.36 pg/mL was found in case 4. PTH amounts of the 2 patients returned to normal 1 year and 1 month post-operation, respectively. Postoperative hematoma, transient recurrent laryngeal nerve paralysis, and infection were not detected.
ATA-HST patient
Only one ATA-HST patient with the RET M918T mutation was found in this study. The female patient presented with the complaint of cervical mass for 7 months. Ultrasound and CT of the cervix revealed multiple goiters in both lobes, with the largest one (3.5 cm × 3.0 cm) located in the right lobe, suppressing the trachea. Enlarged lymph nodes were detected in both lateral cervical compartments. Basal serum calcitonin levels were >2,000 pg/mL; serum CEA amounts were 43.54 ng/mL.
Beside MTC symptoms, the patient had a history of Hirschsprung’s disease and chronic constipation. Mucosal neuromas were found on her lips and tongue. In spite of a young age, development abnormalities, decreased upper to lower body ratio, and Marfanoid habitus were present.
Despite the lack of family history of MTC, the girl was screened for the RET gene and diagnosed as MEN2B. Then, she underwent thyroidectomy and bilateral radical neck dissection. The lesion was found to invade the recurrent laryngeal nerve, vagus, internal jugular vein, and anterior fascia perioperatively. Pathological examination revealed multifocal MTC in bilateral lobes, and LNM was detected in 21/53 lymph nodes. The patient had local lesion recurrence at 34 months postoperatively, and underwent a second surgery; remote metastases occurred 4 months thereafter.
Discussion
In 1985, Takahashi et al. (4) discovered the RET oncogene. In less than a decade, it was demonstrated that virtually all MEN2 patients display RET germline mutations, with approximately 50% of sporadic MTCs having somatic RET mutations. To date, over 100 mutations, duplications, insertions, or deletions involving the RET gene have been identified in patients with hereditary MTC (5-10). In the 2009 ATA guidelines, hereditary MTCs were categorized into A, B, C and D groups, according to increasing aggressiveness. In order to eliminate confusion of hereditary MTC risk categories with other workgroups, e.g. the Seventh International Workshop on MEN, the North American Neuroendocrine Tumor Society, and the National Comprehensive Cancer Network, the new ATA MTC guidelines recommend to change category D to a new category, namely “the highest risk” (HST), category C to “high risk” (H); and A and B to “moderate risk’’ (MOD).
Several studies had shown that distinct RET mutations are always associated with MTC aggressiveness. Studies carried out before clinical genetic screening reported that in four large classical MEN2A families with a RET codon 634 mutation, the average life span of gene carriers was 48 years vs. 60 years in FMTC variant families with a codon 618 mutation (11,12). It was reported that age at initial MTC diagnosis is also correlated with the genotype. The age at conversion from normal to elevated plasma calcitonin levels during a C-cell stimulation test was 18 to 31 years (mean, 23 years) in FMTC variant families, compared with 6 to 33 years (mean, 16 years) in classical MEN2 families (11).
The American M.D. Anderson Cancer Center (13) reported the data of 71 patients from 39 families. Multivariate analysis revealed that the odds of having stage III or IV MTC at the time of diagnosis were increased by 3-fold for each incremental increase in the MTC risk group, from levels 1 to 3. In the Chinese population, the likelihood of ATA-H patients having stage III or IV MTC at the time of diagnosis was 7.888 times that of ATA-MOD patients, as shown above. Thus, in Chinese MEN2A patients there is a correlation between genotype and phenotype, which fits the ATA risk category.
Another high risk factor is age at initial treatment. The probability of advanced MTC increases by 11.6% per year of age at initial treatment, in agreement with M.D. Anderson’s findings. This is likely because nearly 100% of mutation gene carriers would develop into MTC finally. The older the carrier at treatment initiation, the more likely the C cell development into CCH (14), MTC, and advanced disease ultimately. It is worth noting that a C618R mutation carrier underwent surgery at the age of 16, and was diagnosed with multiple focal MTC and bilateral LNM; 1 year later the patient was diagnosed with multiple distant metastasis in the lungs and liver (15). These findings suggested that early treatment is essential, even for patients categorized as ATA-MOD.
When DNA sequencing was unavailable, basal and induced serum calcitonin levels were used to screen potential mutation carriers. Consequently, a large number of patients with no mutations were operated. With the technological development, the importance of calcitonin stimulation test has been reduced considerably (12). The earliest onset ages of 634 and 918 codon mutations are 9 and 2 months, respectively (16,17). Therefore, the ATA guidelines recommended ATA-HST carriers to be operated within the 1st year of life, and ATA-H carriers at the age of 5 years. Prophylactic thyroidectomy could be considered from the age of 5 for ATA-MODs.
However, hypoparathyroidism occurrence after thyroidectomy is significantly higher in very young children. It is difficult to identify the parathyroid glands which are small, translucent, and not fully developed, from the surrounding tissues (18,19). Once hypoparathyroidism occurs, the pediatric patient may suffer from developmental retardation both mentally and physically (20). Therefore, prophylactic thyroidectomy is not recommended to individuals below 2 years of age. In the current study, the surgeon had extensive clinical experience, and used magnifying glasses during operation. In this population, 2 kids developed temporary hypoparathyroidism after thyroidectomy. Parathyroid function in case 1, whose parathyroid was partly or completely embedded in the thyroid, did not recover until a year later.
We believe the current findings may help define a balance point for judging the safety and effectiveness of prophylactic thyroidectomy. All 4 patients in this study underwent surgery only when basal serum calcitonin levels were higher than normal values (21). Only case 1 met the recommendations of the new ATA guidelines. However, all patients were biochemically cured, as assessed at last follow-up. Therefore, delaying prophylactic thyroidectomy until basal serum calcitonin levels are higher than normal values could achieve the same therapeutic effects; meanwhile, the risk of developing hypoparathyroidism would be controlled as well.
We recommend that RET mutation carriers of the ATA-H and ATA-MOD groups should be clinically screened as early as possible based on the ATA guidelines. Surgery is indicated for hereditary MTC patients with positive calcitonin or abnormal thyroid ultrasound results to ensure early cure.
Hereditary MTC with the germline M918T mutation are much more aggressive than MEN2A MTC, and is difficultly cured. A long-term study published in 1985 found that 50% of patients with MEN2B die of MTC, while MEN2A patients show a mortality rate of 9.7% (22). Meanwhile, 95% of MEN2B patients carry the M918T mutation in exon 16. Approximately 75% of MEN2B cases are sporadic and affected patients have de novo RET mutations, while 25% of cases occur in families with previous or current MEN2B manifestations (23,24). This is likely because MEN2B is a highly fatal disease; the patients often present MTC in infancy as well as early metastasis to regional lymph nodes and beyond.
The patient with M918T mutation recruited in this study showed sporadic medullary carcinoma, and her tested parents harbored no mutation. The 12-year-old girl presented advanced disease at initial diagnosis. Local recurrence and distant metastases took place shortly after the initial treatment. The manifestations of this patient were not observed in the MEN2A group. These findings suggested that Chinese MEN2B presents a more aggressive hereditary MTC than MEN2A.
Conclusions
In the Chinese population, the aggressiveness of hereditary MTC is related to specific RET mutations. The risk classification of Chinese hereditary MTC is in line with the new ATA guidelines. We recommend germline RET gene mutation carriers classified as ATA-H and ATA-MOD to undergo close follow up at an early age, with prophylactic thyroidectomy unless basal serum calcitonin is beyond normal.
Acknowledgements
This study was supported by the Capital Health Research and Development of Special (No. 2014-2-026).
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Wells SA Jr, Asa SL, Dralle H, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015;25:567–610. [PubMed] DOI:10.1089/thy.2014.0335
- American Thyroid Association Guidelines Task Force, Kloos RT, Eng C, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 2009;19:565–612. [PubMed] DOI:10.1089/thy.2008.0403
- Wang J, Zhang B, Liu W, et al. Screening of RET gene mutations in Chinese patients with medullary thyroid carcinoma and their relatives . Fam Cancer 2016;15:99–104. [PubMed] DOI:10.1007/s10689-015-9828-6
- Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 1985;42:581–8. [PubMed]
- Donis-Keller H, Dou S, Chi D, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet 1993;2:851–6. [PubMed]
- Mulligan LM, Kwok JB, Healey CS, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A . Nature 1993;363:458–60. [PubMed] DOI:10.1038/363458a0
- Carlson KM, Dou S, Chi D, et al. Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B . Proc Natl Acad Sci U S A 1994;91:1579–83. [PubMed]
- Hofstra RM, Landsvater RM, Ceccherini I, et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma . Nature 1994;367:375–6. [PubMed] DOI:10.1038/367375a0
- Eng C, Smith DP, Mulligan LM, et al. Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours . Hum Mol Genet 1994;3:237–41. [PubMed]
- Marsh DJ, Learoyd DL, Andrew SD, et al. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinoma . Clin Endocrinol (Oxf) 1996;44:249–57. [PubMed]
- Moers AM, Landsvater RM, Schaap C, et al. Familial medullary thyroid carcinoma: not a distinct entity? Genotype-phenotype correlation in a large family. Am J Med 1996;101:635–41. [PubMed]
- Lips CJ, Landsvater RM, Höppener JW, et al. Clinical screening as compared with DNA analysis in families with multiple endocrine neoplasia type 2A. N Engl J Med 1994;331:828–35. [PubMed] DOI:10.1056/NEJM199409293311302
- Yip L, Cote GJ, Shapiro SE, et al. Multiple endocrine neoplasia type 2: evaluation of the genotype-phenotype relationship. Arch Surg 2003;138:409–16. [PubMed] DOI:10.1001/archsurg.138.4.409
- Wolfe HJ, Melvin KE, Cervi-Skinner SJ, et al. C-cell hyperplasia preceding medullary thyroid carcinoma. N Engl J Med 1973;289:437–41. [PubMed] DOI:10.1056/NEJM197308302890901
- Zhang ZX, Li ZJ, Tang PZ, et al. Surgical treatment and prognosis analysis on medullary thyroid carcinoma. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi (in Chinese) 2011;46:209–13. [PubMed]
- Margraf RL, Crockett DK, Krautscheid PM, et al. Multiple endocrine neoplasia type 2 RET protooncogene database: repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations . Hum Mutat 2009;30:548–56. [PubMed] DOI:10.1002/humu.20928
- de Groot JW, Links TP, Plukker JT, et al. RET as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumors . Endocr Rev 2006;27:535–60. [PubMed] DOI:10.1210/er.2006-0017
- Sosa JA, Tuggle CT, Wang TS, et al. Clinical and economic outcomes of thyroid and parathyroid surgery in children. J Clin Endocrinol Metab 2008;93:3058–65. [PubMed] DOI:10.1210/jc.2008-0660
- Tuggle CT, Roman SA, Wang TS, et al. Pediatric endocrine surgery: who is operating on our children?. Surgery 2008;144:869–77. [PubMed] DOI:10.1016/j.surg.2008.08.033
- Frank-Raue K, Buhr H, Dralle H, et al. Long-term outcome in 46 gene carriers of hereditary medullary thyroid carcinoma after prophylactic thyroidectomy: impact of individual RET genotype . Eur J Endocrinol 2006;155:229–36. [PubMed] DOI:10.1530/eje.1.02216
- Elisei R, Romei C. Calcitonin estimation in patients with nodular goiter and its significance for early detection of MTC: European comments to the guidelines of the American Thyroid Association. Thyroid Res 2013;6(Suppl1):S2. [PubMed] DOI:10.1186/1756-6614-6-S1-S2
- Eng C, Clayton D, Schuffenecker I, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis . JAMA 1996;276:1575–9. [PubMed]
- Jasim S, Ying AK, Waguespack SG, et al. Multiple endocrine neoplasia type 2B with a RET proto-oncogene A883F mutation displays a more indolent form of medullary thyroid carcinoma compared with a RET M918T mutation . Thyroid 2011;21:189–92. [PubMed] DOI:10.1089/thy.2010.0328
- Brauckhoff M, Machens A, Lorenz K, et al. Surgical curability of medullary thyroid cancer in multiple endocrine neoplasia 2B: a changing perspective. Ann Surg 2014;259:800–6. [PubMed] DOI:10.1097/SLA.0b013e3182a6f43a