
Molecular genetics and immunohistochemistry characterization of uncommon and recently described renal cell carcinomas
Introduction
Renal cell carcinomas (RCCs) comprise a variety of distinct clinicopathologic entities, each of which displays different morphological, immunohistochemical and molecular features. Landmark consensus conferences held in Heidelberg (1996) and Rochester (1997) provided the basis for much of the last World Health Organization (WHO) renal tumor classification, which appeared in 2004. In the past decade, advances in our understanding of basic morphology, immunohistochemistry, and molecular pathology have led to an expansion in the number of distinct tumor entities that we currently recognize. In 2012, the International Society of Urological Pathology (ISUP) met in Vancouver, BC. The member participants reviewed all aspects of the pathology of adult renal tumors and yielded recommendations relating to proposed new epithelial neoplasms and emerging/provisional new entities. Selected uncommon and recently proposed or emerging new forms of RCC will be discussed in this paper emphasizing on an update of immunohistochemistry, and molecular pathology.
Clear cell (tubulo) papillary RCC (CCP-RCC)
CCP-RCC, also known as clear cell tubulopapillary RCC, is a type of RCC associated with end-stage renal disease (ESRD). Clinically, it was initially described in ESRD by Tickoo et al. (1); however, most cases have been subsequently reported as sporadic (2-9). Indeed, tumors with several overlapping features of CCP-RCC, but with prominent smooth muscle stroma, were reported by Michal et al. 6 years earlier before the report of Tickoo et al. as a disease entity of renal angiomyoadenomatous tumor (1,9-11). Additionally, tumors with a morphology and immune profile similar to CCP-RCC have been reported in von Hippel-Lindau disease (VHL) (12-14). In one study, CCP-RCC-like tumors arising in patients with VHL disease morphologically mimic true CCP-RCC, but the immunohistochemical and genetic features significantly resembled those of clear cell RCC (12).
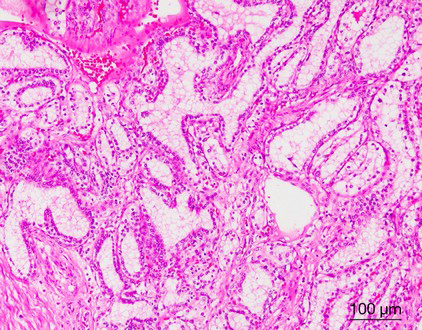
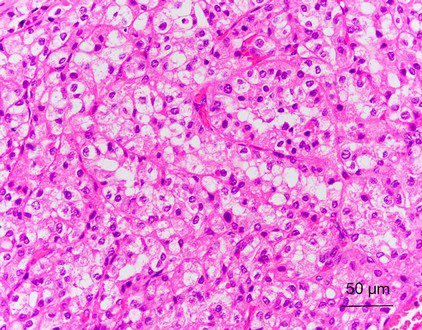
CCP-RCC comprises ~1% of all renal cell neoplasms (1,3). The age of patients with CCP-RCC ranges from 18 to 88 years, with a mean age of 60 years (3-5,15). Grossly, CCP-RCC is generally well defined and well encapsulated. Cystic change or cystic formation are very frequent. The tumors are usually small, solitary, and unilateral, but multifocality and bilaterality have been reported in some cases. The cut surface of the tumor shows a tan-white, pink-tan, yellow, or red-brown color. Necrosis is absent, but focal hemorrhage can be present (1-7). Morphologically, CCP-RCC is composed of various proportions of papillary, tubular/acinar, cystic and solid sheet-like or nested architectures with clear cytoplasm. The papillae are covered by small to medium-sized cuboidal cells and sometimes show extensive secondary branching, which are often folded and densely packed, resulting in a solid appearance. Small blunt papillae, focal branching papillae/acini and interconnecting ribbons are common findings. The nuclei of most CCP-RCCs have a horizontally linear arrangement apart from the basement membrane. The nuclei are round and uniform in shape; nucleoli were not prominent (Fuhrman grade 2) (Figure 1). Foci of large cells or eosinophilic cells can be present. Neither mitotic figures nor necrosis are present (1-7). In many cases, the stroma demonstrates smooth muscle metaplasia that usually connects to the tumor capsule (11). Fibrous stroma is also common (4). Neither foamy macrophages within papillae nor psammoma bodies are present in any tumor.
Immunohistochemically, CCP-RCC expresses cytokeratin 7, hypoxia-inducible factor 1-α (HIF1A) and carbonic anhydrase IX (CA9), but does not express AMACR, CD10, or the transcription factor E3 (TFE3) (2-7). In the study of Leroy et al., cyclin D1 is overexpressed in 83% (35/42) of CCP-RCC cases, so staining for cyclin D1, in combination with CD10, CK7 and AMACR, might be useful in the differential diagnosis from conventional papillary RCC (16).
Molecularly, CCP-RCCs lack deletions of 3p25, VHL gene mutations, VHL promoter hypermethylation, or trisomies of chromosomes 7 and 17 (2,4). Although the mechanism is not yet clear, VHL transcripts are under expressed (5). In clear cell RCC, the VHL gene product protein is important for the regulation of HIF1A and vascular endothelial growth factor (VEGF) expression, whereas loss of function of the VHL gene ultimately leads to overexpression of various proteins that are targets of the HIF pathway, including HIF1A, glucose transporter-1 (GLUT1), and CA9 (5). The strong expression of HIF1A and CA9 in CCP-RCCs provides supporting evidence that up regulation of the HIF pathway in CCP-RCC is independent of VHL gene mutations (5,7). Other events, if any, such as post transcriptional deregulation, translational control or micro-RNA (miR) deregulation, could be a possible explanation for the mechanism of loss-of-function of the VHL gene. In a study of miR expression profiling by Munari et al., a total of 15 cases diagnosed as CCP-RCC were used (17). Among the most differentially expressed miR in CCP-RCC, the authors found that miR-210, miR-122, miR-34a, miR-21, miR-34b*, and miR-489 were upregulated, whereas miR-4284, miR-1202, miR-135a, miR-1973, and miR-204 were downregulated, compared with normal renal parenchyma (17). Furthermore, several studies have documented miR-210 to be overexpressed in clear cell RCC and CCP-RCC (18). MiR-210 is a well-known target of HIF and a central player in the hypoxia pathway (19). A recent report also demonstrated that miR-210 expression positively modulates HIF and correlates with CA9 expression (20). These findings of miR-210 upregulation in CCP-RCC might implicate miR-210 in the activation of the hypoxia pathway in the absence of VHL; however, such a hypothesis would require verification.
A targeted next-generation sequencing and non-coding RNA expression analysis of CCP-RCC by Lawrie et al. found that the miR-200 family is upregulated in CCP-RCC and associated with an unusual epithelial mesenchymal transition (EMT)-marker immunohistochemical staining pattern (21). In their study, all five members of the miR-200 family (miR-200a, miR-200b, miR-200c, miR-141 and miR-429) were significantly upregulated in CCP-RCC cases compared to either clear cell RCC or papillary RCC cases or control tissues. As these miRNAs are intimately involved in the EMT, they stained tissues from CCP-RCC cases for E-cadherin, vimentin (VIM) and β-catenin and found that tumor tissues from all cases were positive for all three markers, a combination rarely reported in other renal tumors that could have diagnostic implications. These data suggest that EMT in CCP-RCC tumor cells is incomplete or blocked, consistent with the indolent clinical course typical of this malignancy (21).
Other studies have demonstrated a relationship between CCP-RCC and other RCC. The miR expression profiling study by Munari et al. revealed that the miR expression profile of CCP-RCC more closely resembled clear cell RCC for upregulated miRs and papillary RCC for downregulated miRs (17). Additionally, as expected, the CCP-RCC miRNA profile more closely resembled primary rather than metastatic clear cell RCC. In a gene expression profiling study of CCP-RCC by Fisher et al., compared with papillary RCC, CCP-RCC expressed more CA9, CP, NNMT, and VIM; less AMACR, BAMBI, and SLC34A2; and similar levels of SCHIP1. Compared with clear cell RCC, CCP-RCC expressed slightly less NNMT, but similar levels of the seven other genes (22). Although CCP-RCC exhibits a unique molecular signature, it expresses several genes at comparable levels to clear cell RCC relative to papillary RCC.
Additionally, a comparative genomic hybridization study of Adam et al. demonstrated no genomic imbalance in the seven tumors tested, whereas other scattered reports using various methods have shown abnormalities at various chromosomal loci, including trisomies 10 and 12; monosomies 3, 16, 17, and 20; and gains at 5p, 5q, 7pq, 12pq, and 16pq (4,7,23-25). Understanding the molecular pathogenesis of CCP-RCC will play a key role in the future subclassification of this unique tumor.
MiTF family translocation RCC
The microphthalmia-associated transcription factor (MiTF) subfamily of transcription factors includes TFE3, TFEB, TFEC, and MiTF. All family members share a homologous basic helix-loop-helix DNA binding domain and have overlapping transcriptional targets (26). Several distinct tumors are associated with the overexpression of this gene family, including translocation-associated RCCs, alveolar soft part sarcoma, melanoma, clear cell sarcoma, angiomyolipoma, and perivascular epithelioid cell neoplasms (PEComas). All these tumors have been considered to be members of the family of tumors, owing to their histological, immunohistochemical and molecular genetic similarities (26,27).
Xp11 Translocation RCC
RCCs associated with Xp11 translocation are uncommon renal tumors, which were recognized as a distinct entity in the 2004 WHO classification system. They are characterized by several different translocations that involve chromosome Xp11, resulting in gene fusions of the TFE3 gene (27). In these diseases, the TFE3 gene is fused via translocation to one of several other genes, including ASPL, PRCC, NONO (P54NRB), CLTC, PSF, LUC7L3, KHSRP, and unknown genes on chromosomes 3, 10 and 19. The most common translocations are ASPL-TFE3 and PRCC-TFE3 fusions (27-29). Of interest, both ASPL-TFE3 RCC and alveolar soft part sarcoma (ASPS) harbor the same ASPL-TFE3 fusion gene. However, the translocation is balanced in ASPL-TFE3 RCC and is unbalanced in ASPS. The function of chimeric TFE3 fusion proteins can also vary. In the study by Tsuda et al., ASPL-TFE3, PSF-TFE3 and NONO-TFE3 all bind to the MET promoter. However, ASPL-TFE3 induces a much stronger upregulation of downstream MET receptor tyrosine kinase than does PSF-TFE3 or NONO-TFE3 (30). Evidence suggests that the chimeric PRCC-TFE3 and NONO-TFE3 are more potent as transcription factors than wild-type TFE3, while PSF-TFE3 and CLTC-TFE3 can interfere with cell cycle control (30,31).
Xp11 translocation RCC is estimated to represent 33% to 50% of pediatric RCC and <1% to 4% of adult RCC (28,32-34). In children, studies have indicated that previous chemotherapy might be a risk factor for developing Xp11 translocation RCC (35). Up to 15% of patients with Xp11 translocation RCC have had a history of chemotherapy exposure (35). In adults, although Xp11 translocation RCC has been reported during pregnancy or in association with ESRD and hemodialysis, no studies have been carried out to identify particular risk factors (36,37).
From a clinical outcome perspective, outcome data on Xp11 translocation RCC remain at a premature stage because of its relatively rare incidence. Several studies have demonstrated that Xp11 translocation RCC have a relatively indolent course, despite their often advanced stage at presentation (38). However, some cases of Xp11 translocation RCC have been previously reported to follow an aggressive clinical course (28,39-41). Late recurrences, as many as 17 or 30 years after original diagnosis, have been interpreted to suggest the tendency of these RCCs to recur late (42,43). Unfortunately, Xp11 translocation RCCs in adults often present with advanced disease and distant metastases and a worse prognosis (28). Satisfactory long-term follow up data are necessary before any favorable short-term outcome can be confirmed. Targeted therapies, such as VEGF receptor-targeted agents and mammalian target of rapamycin (mTOR) inhibitors, have been increasingly used to treat metastatic RCC, particularly clear cell RCC. However, recent studies have found that such agents might also be beneficial in patients with Xp11 translocation RCC (44,45). Therefore, discriminating Xp11 translocation RCC from other subtypes of renal tumor is clinically important.
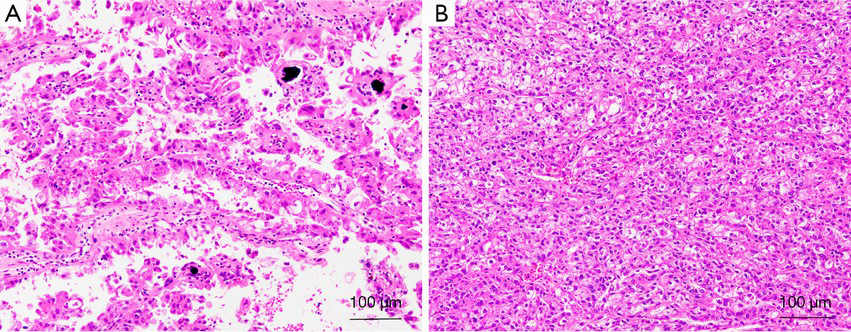
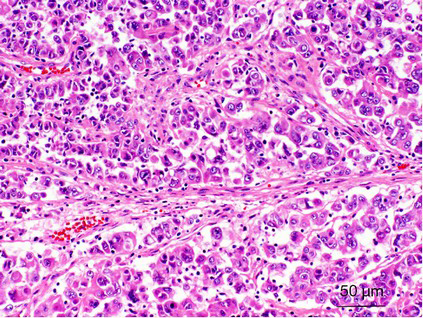
Morphologically, most of these tumors characteristically have a typical morphology, including alveolar, nested, or papillary architecture, with discrete cell borders, voluminous cytoplasm, prominent nucleoli, and psammoma bodies (Figure 2). However, there has been increasing evidence that MiTF/TFE renal translocation carcinomas might demonstrate unusual morphological features that mimic other types of RCCs, including clear cell RCC, multilocular cystic RCC, pleomorphic giant cells, collecting duct carcinoma, high-grade urothelial carcinoma and well-developed fascicles of spindled neoplastic cells with bland nuclei and focal myxoid stroma (28,41,46,47).
Unlike other types of RCC, Xp11 translocation RCCs are negative or only focally positive for cytokeratins and epithelial membrane antigen (EMA) and have variable VIM expression. CK7 is typically negative. These tumors characteristically express CD10, the RCC marker, and racemase (AMACR; P504S), and most express PAX2 and PAX8. Some Xp11 translocation RCCs with a typical morphology express melanocytic markers, such as Melan-A and HMB-45 (28,41,46,47).
Cathepsin K is a cysteine protease of the papain family, which plays an important role in osteoclast function. Expression of cathepsin K in osteoclasts is regulated by MITF. As recent studies have demonstrated cathepsin K to be a transcriptional target of the MITF family, an immunohistochemistry antibody to cathepsin K has been utilized in the diagnosis of MiTF family translocation RCC (48). Cathepsin K is overexpressed in a subset of Xp11 translocation RCCs and in almost all t(6;11) RCCs, but not in other types of RCCs. Martignoni et al. found cathepsin K to be differentially expressed in a manner dependent upon the fusion partner of the TFE3 gene. In that study, 12 of 14 Xp11 RCCs with PRCC-TFE3 gene fusion showed a positive reaction with antibody to cathepsin K, whereas all 8 carcinomas with ASPSCR1-TFE3 gene fusion showed negative staining, suggesting that there are functional differences between the resulting fusion proteins (49).
The most sensitive and specific marker for the Xp11 translocation RCC is strong nuclear TFE3 immunoreactivity, using an antibody to the C-terminal portion of TFE3. However, results can vary between laboratories because of technical factors, such as tissue preservation, fixation time, antigen retrieval methodology, sensitivity and specificity of the antibody used, cutoff point, and data interpretation. There has been increasing evidence that TFE3 immunohistochemistry can yield false-positive, false-negative, or equivocal results (46,50-53). Cytogenetic karyotypic analysis and reverse transcriptase polymerase chain reaction are both common molecular methodologies for identifying this translocation. Unfortunately, these methods can be limited by the availability of viable tumor cells, unfixed tissue, or other special handling techniques, which are not always incorporated into routine surgical pathology practices. Recently, several TFE3 break-apart FISH assays have been developed to detect TFE3 gene rearrangements. The TFE3 break-apart FISH assay is a useful complementary method for confirming a diagnosis of Xp11 translocation RCC, especially when the morphological or clinical suspicion is high, but TFE3 immunostaining is negative or equivocal (46,47).
Xp11 neoplasm with melanocytic differentiation
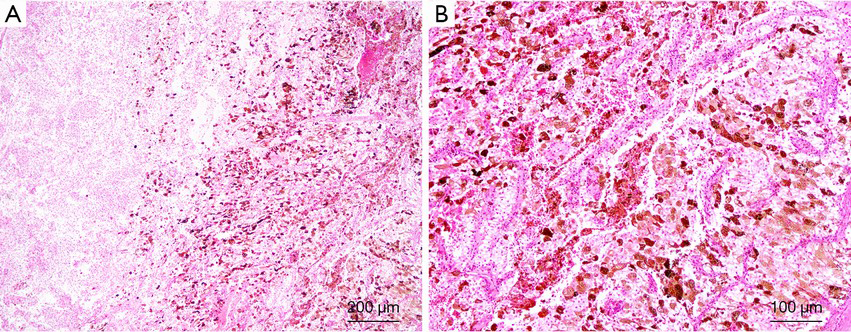
Recently, an increasing number of TFE3 rearrangement-associated tumors with melanocytic differentiation have been reported, such as TFE3 rearrangement-associated perivascular epithelioid cell tumors (PEComas), melanotic Xp11 translocation renal cancers, and melanotic Xp11 neoplasms (54). These tumors displayed purely nested or sheet-like architectures separated by delicate vascular networks and were predominantly composed of purely epithelioid cells containing a clear or granular eosinophilic cytoplasm (Figure 3). By immunohistochemistry, the tumor cells were immunoreactive for the melanocytic markers HMB45 and Melan A but not for cytokeratins, muscle markers, S100, and the renal tubule markers CD10, PAX2, and PAX8 (54). Argani et al. suggested that these tumors have a phenotype that most closely resembles PEComa (27). However, PSF/SFPQ has been found to be a very common gene fusion partner in TFE3 rearrangement-associated PEComas, melanotic Xp11 translocation renal cancers, and melanotic Xp11 neoplasms (54-56). Considering the shared clinicopathologic characteristics (young adult and female predominance), morphology, immunophenotypic characteristics, genetic alterations (TFE3 gene fusion) and uncertain clinical behavior that sometimes follows an aggressive course, we and others have suggested that they belong to a single clinicopathologic spectrum. “Xp11 neoplasm with melanocytic differentiation” or “melanotic Xp11 neoplasm” has been proposed to designate this unique neoplasm (54,57).
t(6;11) RCC
Another rare subset of renal translocation tumors is associated with t(6;11)(p21;q12), which results in an Alpha-TFEB gene fusion. The distinctive morphological, immunohistochemical, ultrastructural, and cytogenetic features of this neoplasm were first described by Argani et al. in 2001 (58). To the best of our knowledge, fewer than 60 cases have been described in the literature (58-71). These tumors mainly affect children and young adults. Information regarding the clinical behavior of TFEB RCCs has remained uncertain because of their rare incidence. TFEB RCCs seems to have a relatively indolent course, despite recurrence in 17% of patients, some cases presenting with metastasis, and the occasional selection of an aggressive clinical course that causes death (50,62,66).
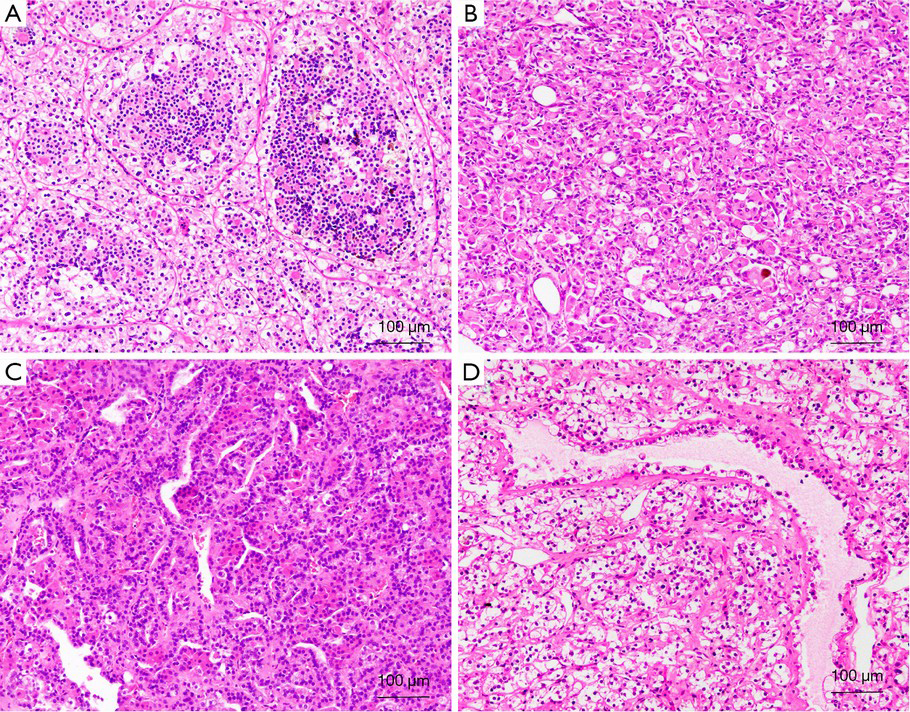
Morphologically, the tumors characteristically have nests or sheets of epithelioid cell morphology with clear cytoplasm, mimicking a typical clear cell RCC, and clusters of small cells that are usually clustered around hyaline material (Figure 4A). In some tumors, pigment and psammoma bodies can be observed. However, there has been increasing evidence that the t(6;11) RCC might demonstrate unusual morphological features, such as epithelioid angiomyolipoma structures (Figure 4B), papillary (Figure 4C), tubular, chromophobe cell RCC, and clear cell RCC (Figure 4D) (68-71).
Immunohistochemically, the tumors express melanocytic immunohistochemistry markers, such as HMB-45 and Melan-A, but are either negative or only focally positive for epithelial markers, such as cytokeratins. Cathepsin K is overexpressed in almost all t(6;11) RCCs (48). Most cases express PAX8, supporting renal tubular differentiation (68-71). As a result of a promoter substitution, the Alpha-TFEB gene fusion results in the overexpression of native TFEB in t(6;11) RCC. A recently developed antibody that recognizes TFEB has been shown to be a surrogate marker for t(6;11) RCC (61). However, these results can vary or be equivocal between laboratories because of different technical differences (68). Additionally, the Alpha-TFEB fusion point seems to vary from case to case (68). Thus, the reverse transcriptase polymerase chain reaction to detect fusion genes is not always easy to apply in routine diagnostic practice. Recently, a break-apart FISH assay for TFEB gene fusions has been developed for archival material and has allowed for the expansion of the clinical and morphological spectrum of t(6;11) RCC (68,69).
Succinate dehydrogenase (SDH)-deficient RCC
The SDH enzyme complex is located in the inner mitochondrial membrane, and it is a mitochondrial enzyme complex that participates in the electron transport chain (complex II) and tricarboxylic acid cycle (Krebs cycle) by catalyzing the oxidative dehydrogenation of succinate to fumarate. The complex consists of four different subunit proteins (SDHA, SDHB, SDHC, and SDHD), as well as assembly factors (SDHAF1 and SDHAF2), iron-sulphur centers, and ubiquinone (72,73). Germline mutations of the genes that encode the SDH subunits can result in hereditary paraganglioma pheochromocytoma syndromes, as well as gastrointestinal stromal tumors (GISTs) that can be recognized by a distinctive multinodular architecture, and predominantly exhibit an epithelioid morphology and a predilection for lymph node metastasis (72-74). In addition to paraganglioma and GIST, increasing evidence suggests that patients with germline mutations of SDH subunit genes also develop renal tumors (75). The first SDHB-related RCCs were described in two families with a history of paraganglioma, RCC, germline SDHB mutations, and loss of heterozygosity for the region encoding SDHB in their tumors tissue in 2004 (76). Subsequently, less than 78 cases having been described in detail (76-81).
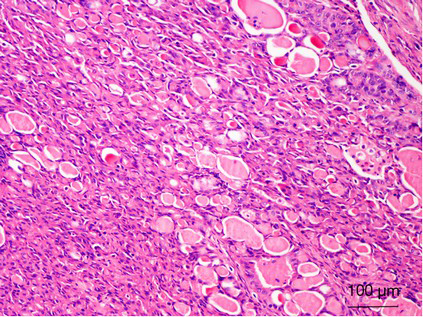
SDH-deficient RCCs are commonly multifocal; with prolonged follow-up, bilateral tumors can be identified in up to 26% of patients (80). To date, nearly all reported cases have been associated with germline mutations of the SDH genes (80,81). Histologically, most of them exhibit a distinctive morphology that can be distinguished from other subtypes of renal tumors. These tumors have been observed to be formed by a uniform population of cells with an eosinophilic cytoplasm and cytoplasmic inclusions of flocculent or eosinophilic material (previously reported to be abnormal mitochondria), and they exhibit a predominantly solid or nested architecture (Figure 5). Intratumoral mast cells, entrapped nonneoplastic renal tubules, and a variable tumor pseudocapsule are other characteristic features (78,80,81). However, RCCs with other histological appearances, including tumors resembling clear cell RCC, papillary RCC, sarcomatoid and unclassified RCCs, have been reported in patients with germline mutations of SDH subunit genes, and a few RCCs of other histological types have been found to be SDH-deficient in the absence of a known germline gene mutation (78-82). The significance of this finding remains incompletely understood.
By immunohistochemistry, the tumors are consistently negative for SDHB, CD117, RCC antigen and CA9. Most tumors are positive for PAX-8, and kidney-specific cadherin. Immunohistochemical labeling for CD10, cytokeratin AE1/AE3, CAM5.2, CK7, AMACR, and EMA vary widely (80,81). Loss of SDHB in tumors cells reflects dysfunction of the SDH complex, which might be caused by mutations in the genes that encode any of the four subunit proteins, or by deficient complex activity that is secondary to other, as yet unknown, mechanisms. By contrast, loss of SDHA expression can only be observed in tumors with mutations in SDHA (74).
It is likely that most SDH-deficient RCCs will be associated with a SDHB germline mutation, and small subsets have been reported to arise in the settings of SDHA, SDHC or SDHD genes (80,81,83). The extremely high rate of SDHB germline mutations in the SDH subunits in SDH-deficient RCC differs from that found in SDH-deficient GIST, in which ~30% of cases are associated with a SDHA mutation, and 10% to 20% of cases are associated with mutations in other SDH subunits (SDHB, SDHC, or SDHD), and the rest cases without mutations may be correlated with aberrant DNA hypermethylation of SDHC as recently reported (74,80,84,85). Additionally, the targeted next-generation sequencing panel utilized in the study of Williamson et al. did not reveal alterations in other key genes involved in RCC pathogenesis, such as VHL, PIK3CA, AKT, MTOR, MET, or TP53 (81).
The precise mechanisms whereby SDH complex dysfunction leads to tumor formation have not yet been fully elucidated. Based largely on data from in vitro biochemical studies, it was suggested that increased levels of succinate can result in the stabilization of HIF1A, leading to the transcription of target genes, such as the gene encoding VEGF (74,86). However, Fleming et al. investigated three cellular signaling pathways that could be involved in tumorigenesis in SDH-deficient RCCs (87). They found no evidence supporting a role for either the mTOR [p-mTOR (Ser2448), p-S6 riboprotein (Ser235/236)] or hypoxia-inducible (carbonic anhydrase 9 and EGFR) pathways. However, they did detect immunohistochemical reactivity for phosphorylated AMP-dependent kinase (p-AMPK Thr172), glycogen synthase kinase 3 (GSK3) phosphorylation (p-GSK3 Ser12) and nuclear expression of cyclin D1. These findings suggest that failure of ATP generation and activation of AMPK are important steps in the pathogenesis of SDH-deficient RCCs and that these tumors might arise through a mechanism that involves ATP depletion, activation of AMPK, and induction of cyclin D1. This could represent a unique pathway for tumor development that has the potential for therapeutic interventions in these rare tumors.
SDH-deficient RCC represents a distinct and rare renal neoplasm, which is defined by loss of immunohistochemistry staining for SDHB and has been accepted as a provisional entity in the 2013 ISUP Vancouver Classification (33).
ALK Translocation RCC
Chromosomal rearrangements involving the anaplastic lymphoma kinase (ALK) gene at 2p23 were first recognized in anaplastic large cell lymphoma (88). The characteristic translocation was shown to result in fusion of the 3’-portion of ALK with the 5’-portion of NPM1 at 5q35 to produce a chimeric oncogene (89). Subsequently, ALK rearrangements have been found in diffuse large B-cell lymphoma, plasmacytoma, inflammatory myofibroblastic tumors, esophageal squamous cell carcinoma, breast carcinoma, colonic adenocarcinoma, non-small cell lung cancer and, recently, RCC (90-96).
To date, only ~8 cases of ALK translocation RCC have been described by 6 independent research groups (97-103). A total of three of the ALK translocation RCCs occurred in young patients (ages 6, 16, and 6 years old) who were African American males with the sickle-cell trait, and in all cases, the ALK gene was fused to the vinculin (VCL) gene, which functions as an adhesion protein that couples the extracellular matrix to the actin-myosin cytoskeleton (97,98,103). A review of these cases separately reported that VCL-ALK RCCs have many similar and distinctive features, including their occurrence in young patients with the sickle-cell trait, medullary epicenter, solid architecture, distinctive polygonal to spindle-shaped cells with vesicular nuclei and abundant eosinophilic cytoplasm with prominent intracytoplasmic lumina, prominent lymphoplasmacytic infiltrate, intact INI1 protein, and a low Ki-67 index (97,98,103).
Given the shared clinicopathological characterizations with renal medullary carcinoma (RMC), it has been proposed that VCL-ALK RCC might represent a subtype of RMC. However, several features distinguish VCL-ALK RCC from classic RMC. Similar to rhabdoid tumors and a subset of collecting duct carcinomas, RMC typically demonstrates loss of the INI1 protein by immunohistochemistry, a finding almost never observed in the more common RCC subtypes, whereas INI1 protein in VCL-ALK RCC remains intact. Furthermore, RMCs are high-grade cancers that are characterized by an extremely high mitotic rate and Ki-67 proliferation index, whereas the proliferative activity of VCL-ALK RCC is relatively low. Given the very small number of these RCCs that have been reported, the relationship between RMC and VCL-ALK RCC is not yet understood.
Subsequently, 5 cases of RCC associated with ALK rearrangements have been reported in older adults (ages 36, 53, 61, 59, and 44 years old) (99-102). Among those patients, two Japanese cases described by Sugawara et al. were in adults with different translocations: TPM3-ALK (found in anaplastic large cell lymphoma and inflammatory myofibroblastic tumor) and EML4-ALK (reported to be identified in lung, breast and colon adenocarcinomas) (99). VCL was not the partner in the two cases described by Sukov et al. (100). Fusion partners of ALK were not detected in one Korean case (102). Their morphology has not been distinctive: four of the neoplasms have been classified as variants of papillary RCC, whereas the other was considered RCC unclassified. Such findings are in contrast to those reported for three cases with VCL/ALK fusion. In two cases described by Sukov et al., both patients with ALK rearrangement had a poor clinical course, surviving less than 5 years post-diagnosis (4 years and 1.4 years to death from RCC) (100). Hodge et al. therefore proposed that ALK rearrangement in papillary RCC cases in older adults could represent a distinct pathological entity with poorer outcome (101).
The ALK protein is a membrane-associated tyrosine kinase that belongs to the insulin receptor superfamily. Outside of the embryonic state, ALK expression is normally confined to the central nervous system (89). Chromosomal rearrangements resulting in ALK gene fusion lead to aberrant ALK activation by the formation of chimeric protein products, which exhibit a cytoplasmic and/or membranous distribution, depending on the fusion partner. The ALK fusion products have significant oncogenic activity (104). Because of their expression pattern, ALK fusion products have become an enticing chemotherapeutic target. Recently, the ALK inhibitor crizotinib has been used in patients with lung cancer, inflammatory myofibroblastic tumors (IMTs), or anaplastic large cell lymphomas (ALCLs), which can harbor various ALK fusions (99,105,106). Such distinctions for ALK-rearranged RCCs are of particular importance and should be recognized because of the potential benefit of ALK inhibitor therapy in these patients.
Carcinoma associated with ESRD
Acquired cystic disease–associated RCC
The relationship between ESRD and the development of renal neoplasia has been increasingly recognized in the past decade. The spectrum of renal tumors associated with ESRD is quite varied. In single cases and small series of clear cell RCCs, papillary RCCs, chromophobe RCC, collecting-duct carcinoma, tubulocystic carcinoma, angiomyolipoma, oncocytoma and mixed epithelial and stromal tumor have been reported (107-111). In addition to recognized forms of renal neoplasia, recent studies have identified two distinct renal-cell neoplasms with unusual histological patterns that do not fit into the categories recognized in the current classification system (1,111).
One group of these RCCs by definition is associated with features of end-stage kidney disease with acquired renal cysts in the background kidney, which is designated acquired cystic disease-associated RCC. Acquired cystic disease–associated RCC is the most common subtype of RCC that is present in end-stage kidneys, accounts for 36% of RCCs present in end-stage kidneys, and is present in 46% of kidneys with acquired cystic disease (1). Because most tumors are diagnosed incidentally and early on for long-term radiologic follow-up for chronic renal disease, acquired cystic disease–associated RCC has a relatively good prognosis (1); these tumors are usually well circumscribed, and they can be surrounded by a thick fibrous capsule that shows dystrophic calcification. However, some rare typical cases, as well as tumors with sarcomatoid or rhabdoid features, can metastasize (1,112).
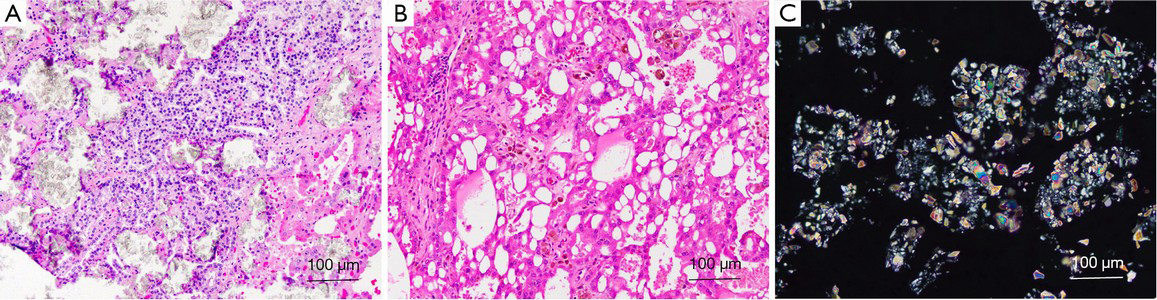
Microscopically, there are a variety of architectural patterns, with solid, acinar, cystic and papillary patterns being present (Figure 6A). Most tumor cells have abundant granular eosinophilic cytoplasm, with round to oval nuclei that exhibit vesicular chromatin and prominent nucleoli (Figure 6B). Foci with a clear to vacuolated cytoplasm can also be present. In 67% of reported cases, the tumor appeared to arise in a cyst (111). A cribriform/sieve-like appearance, prominent nucleoli (high-grade nuclei), and intratumoral oxalate crystal (Figure 6C) deposits are its characteristic features (1,113).
Acquired cystic disease–associated RCCs are diffusely positive for AMACR and, in a proportion of cases, show variable and predominantly focal staining for CK7. Positivity for CD10, RCC, CD57, GST-α, PTEN (phosphatase and tensin homolog deleted on chromosome 10) and c-met have also been described. Staining for EMA, high-molecular-weight cytokeratin, CA-9, CD57, CD68, c-kit, Pax-2, platelet-derived growth factor receptor (PDGFR)-α, and VEGF receptor (VEGFR)-2 are negative (111,114-119).
Genetic studies on acquired cystic disease–associated RCCs are limited. Comparative genomic hybridization (CGH) and fluorescence in situ hybridization (FISH) analyses have revealed gains and losses of multiple chromosomes. Although gains of chromosomes 1, 2, 3, 6, 7, 16, 17, X, and Y were frequently observed, losses of chromosomes 3, 9, and 16 have also been reported (112,115,117,120,121). In addition, gains of chromosome 3 have been among the more consistent findings (115,117,120-122). Mutations of the VHL gene have not yet been identified in these tumors (114).
Hereditary leiomyomatosis RCC syndrome-associated RCC
Hereditary leiomyomatosis RCC is an autosomal-dominant hereditary syndrome that was initially reported in 2001 by Kiuru et al. (123). Individuals affected with this syndrome are at risk of developing cutaneous leiomyomas, multiple symptomatic uterine fibroids in young women, and early onset renal tumors with a type 2 papillary morphology that can progress and metastasize, even when they are small (124,125). Additionally, there have also been case reports of associations with other malignancies, including bladder cancer, adrenocortical tumors, Leydig-cell tumors of the testis, ovarian cystadenomas and gastrointestinal stromal tumors, although the significance is yet to be determined (126). Indeed, hereditary leiomyomatosis RCC-associated RCC has been classified in the 2004 WHO classification of renal neoplasms in the section related to hereditary RCC as a hereditary counterpart of type 2 papillary RCC. However, it is now known that hereditary leiomyomatosis RCC-associated RCC (although usually having a papillary pattern) is very aggressive, prone to metastasis, and lethal if allowed to progress compared with renal tumors in other hereditary renal cancer syndromes and, as such, should be recognized as a distinctive tumor subtype (125,127,128).
Unlike other familial RCC syndromes that can typically develop multiple and bilateral tumors, RCCs in the hereditary leiomyomatosis RCC setting can be solitary and unilateral, and not much is known with regards to their hereditary nature. Morphologically, hereditary leiomyomatosis RCC-associated RCC can have a type 2 papillary morphology or a solid architecture. However, the histopathological spectrum of these tumors has recently expanded and includes architectural patterns that have papillary, tubulopapillary, tubular, solid, and cystic sarcomatoid rhabdoid elements (129,130). Some cases histologically resemble collecting duct carcinoma, tubulocystic carcinoma or clear cell RCC (131-133). The seminal work by Merino et al. proposed that the morphologic hallmarks of hereditary leiomyomatosis RCC-associated RCC are the following characteristic nuclear features: large nuclei, prominent inclusion-like eosinophilic nucleoli, and perinucleolar clearing (Figure 7). This broad morphological spectrum leads to a difficult diagnostic challenge (129). Immunohistochemistry for CK7 and CK20 and high-molecular-weight cytokeratins (i.e., CK1, CK5, CK10, and CK14) is routinely negative (130).
The gene responsible for hereditary leiomyomatosis RCC was identified by Tomlinson et al. on chromosome 1 (1q42-44), which is known as fumarate hydratase (FH) (124). This gene acts as a tumor suppressor, and both alleles are inactivated in tumor tissue caused by germline mutations in one allele. Frequent loss of heterozygosity occurs for chromosome 1q that harbors the second allele (124). FH encodes an essential enzyme in the Krebs (citric acid) cycle that catalyzes the conversion of fumarate to malate (125). Alteration to FH up regulates HIF and creates a pseudohypoxic environment, which is similarly seen in VHL. When FH is inactivated, fumarate levels build up and competitively inhibit HIF prolyl hydroxylase (HPH). HPH is a key enzymatic regulator of intracellular HIF levels (134). When HPH is inactivated, HIF levels increase, and transcription of downstream genes occurs (125,134). Potential areas of systematic therapy for hereditary leiomyomatosis RCC will likely be designed to prevent increases in HIF levels or target the transcription products of VHL-independent HIF accumulation, such as VEGF and TGFA/EGFR.
On the other hand, the functional loss of FH also leads to the abnormal intracellular accumulation of fumarate. Interestingly, increased levels of fumarate in cells can cause aberrant succination of cellular proteins by forming a stable chemical modification, S-(2-succino)-cysteine (2SC), which can be detected by immunohistochemistry (135-138). In contrast to typical immunohistochemistry assays in which a peptide or protein is the specific antigen, 2SC immunohistochemistry stains all of the proteins with a stable 2SC modification (succination). Therefore, in different cellular contexts, the staining pattern can vary broadly for nuclear, cytoplasmic, and mitochondrial proteins (139,140). Based on this finding, Bardella et al. showed that detecting 2SC positivity by immunohistochemistry can predict the mutational status of the FH gene, and this immunoreactivity was absent in a diverse group of nonhereditary leiomyomatosis RCC-related tumors (139). In another series, Chen demonstrated that immunohistochemical detection of 2SC is a useful ancillary tool in the differentiation of hereditary leiomyomatosis RCC-associated RCC from other high-grade RCCs, and nuclear protein succination could be a feature of hereditary leiomyomatosis RCC tumors (140).
Thyroid-like follicular RCC
Thyroid-like follicular RCC is an unusual histological variant of RCC that was not included in the current WHO classification of renal tumors. Thyroid-like follicular RCC was first described in abstract format by Amin et al. in 2004. To date, 20 cases have been reported in the English literature (141-153). However, because of the limited number of cases and the uncertainty about the maximum allowable papillary component in this tumor, it was only recognized as an emerging entity in the 2013 Vancouver classification of renal neoplasms (33).
Thyroid-like follicular RCC are usually detected in middle-aged patients (mean: 44 years; range, 19–83 years), with 11 women and 9 men affected. The primary tumor sizes range from 1.9 to 11.8 cm with a tan color and are solitary in all cases to date (141-153).
Most thyroid-like follicular RCC followed an indolent course with metastatic disease observed in four patients (142-144,152). One patient developed a lung metastasis 2 months after initial diagnosis (142); a second patient presented with metastatic extension to the renal hilar lymph nodes, and another two patients presented with metastases to the lungs and lymph nodes (143,144,152). These latter cases provide evidence that this rare variant of RCC has a low, but distinct, malignant potential and can be clinically aggressive.
Microscopically, tumors showed a circumscribed nonencapsulated neoplasm with a diffuse follicular growth pattern. The follicles were variable in size, and some were cystically dilated and filled with a pink colloid-like material (Figure 8). They were lined by cells with a moderate amount of amphophilic to eosinophilic cytoplasm. The nuclei are generally round and regular and contain enlarged nucleoli (nucleolar grade 2 or 3). A solitary case without a papillary architecture exhibited a striking resemblance to the nuclear features of papillary thyroid carcinoma, including nuclear membrane irregularities, nuclear grooves, and nuclear clearing that resembles the follicular variant of papillary thyroid carcinoma (141-153). Another special case consisted of two components; the papillary RCC-like and thyroid follicular-like carcinoma areas, accounting for 70% and 30%, respectively. Both areas were intimately intermingled with each other (154).
Before making a definitive diagnosis of thyroid-like follicular RCC, a detailed work-up is needed to exclude the possibility of metastatic follicular carcinoma or a follicular variant of papillary thyroid carcinoma. These neoplasms stain negatively for thyroid transcription factor-1 (TTF1) and thyroglobulin, allowing their distinction from follicular carcinoma or a follicular variant of papillary thyroid carcinoma. Immunohistochemically, the tumors are consistently negative for TTF1 and thyroglobulin, which are markers of thyroid carcinoma. Most tumors are positive for AE1/AE3, CAM5.2, PAX-8, EMA, CK19, CK7, and VIM. Variable expression in thyroid-like follicular RCC can be observed for CK34βE12, PAX-2, CD10, CK20, and AMACR (141-153).
Various genetic abnormalities have been identified in thyroid-like follicular RCC, without any distinctive pattern emerging. Using comparative genomic hybridization analysis, Jung et al. identified losses of chromosomes 1p36, 3 and 9q21-33 and gains of 7q36, 8q24, 12, 16, 17p11-q11, 17q24, 19q, 20q13, 21q22.3, and Xp in one tumor (141). Another tumor demonstrated chromosomal losses of chromosomes 1, 3, 7, 9p21, 12, 17, and X by FISH. These genetic abnormalities are distinct from the ones observed in clear cell or chromophobe RCC. By contrast, no genetic alterations were observed in the third case analyzed using comparative genomic hybridization. In the study of Amin et al., gene expression profiling in 3 tumors showed 135 overexpressed genes and 46 underexpressed genes in thyroid-like follicular RCC compared to clear cell and chromophobe RCC (143). In particular, the expression of the mixed lineage leukemia (MLL) gene was increased 2.5-fold in thyroid-like follicular RCC. The MLL gene encodes a transcriptional factor that is involved in the oncogenesis of some hematologic malignancies, including lymphoblastic lymphomas and leukemias and 8% of T-ALL (155). Notably, three previous thyroid-like follicular RCC patients also had history of hematologic disorders, including Hodgkin lymphoma, acute myeloid leukemia, and T-ALL (145,148,153). Based on these findings, the genes involved in lymphoma and leukemia pathogenesis might play a role in the development of thyroid-like follicular RCC, although additional study is needed.
Tubulocystic TCC
Tubulocystic RCC is an uncommon and recently described variant of RCC, which was not listed in the 2004 WHO classification. The first detailed description of tubulocystic RCC came in 1997 when MacLennan et al. described 13, cases which they classified as low-grade mucinous tubulocystic renal cancer likely of collecting duct origin because of mucin production within a tumor (156). In contrast to the highly aggressive collecting duct carcinomas, these had less aggressive behavior, and only one patient died of metastatic disease (156). Three large series of tubulocystic RCC by Azoulayet al, Yang et al., and Amin et al. adequately confirmed the histological features and documented gene profiling data in these tubulocystic RCC (157-159). Less than 70 cases of renal tubulocystic carcinoma have now been reported, and they occur in men approximately 7 times more often than in women at a mean age of 60 years (range, 18 to 94 years). Most cases are discovered incidentally and are typically small when discovered (nearly 40% are 2 cm or smaller) (157-160).
Tubulocystic RCC is a well-circumscribed tumor that ranges in size from 0.5 to 17 cm, is usually grossly cystic and lacks hemorrhage or necrosis (157-160). Histologically, the tumor is composed of tubules and cystic structures of markedly variable size, separated by septa that are commonly delicate like a spider web or by fibrotic septa of variable thickness. Tubules are lined by a single layer of low cuboidal epithelial cells with modest to abundant amounts of eosinophilic cytoplasm, and they often have, at least focally, a hobnail configuration. (Figure 9) Nuclei are round, with evenly dispersed chromatin, and they have prominent nucleoli (nucleolar grade 3) in most cases. No necrosis is present, and mitoses are inconspicuous (157-160).
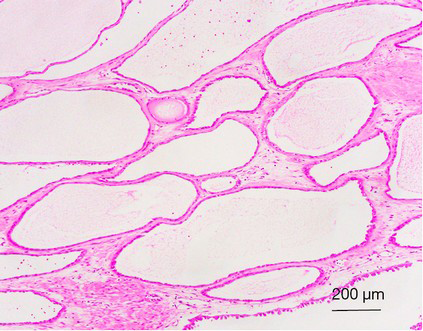
By immunohistochemistry, the tumors are positive for cytokeratins CK8, CK18, and CK19 and nearly always negative for high-molecular-weight cytokeratin (34βE12). CD10 and AMACR (racemase) are positive in greater than 90% of tumors. CK7 is commonly expressed, although the pattern can be weak and focal. Less than half of the cases will be positive for PAX2 and carbonic anhydrase IX (CA-IX). Staining for kidney-specific cadherin can also be investigated (111,157-159).
There is a limited number of molecular studies on tubulocystic RCC because of its rarity. Genetic studies indicate that tubulocystic RCC has a distinct molecular signature (161). A recent study by Amin et al. found that the gene expression profile of 5 tubulocystic RCC cases did not overlap with that of papillary RCC, as determined in the investigators’ earlier studies (159). A comparative study of the gene expression profiles of tubulocystic RCC and collecting-duct carcinoma showed significant differences, indicating that these two tumor types are unrelated (162). However, in some studies, it has been suggested that tubulocystic RCC and papillary RCC show some similarities to each other in both genetic changes and immunoexpression. A gene expression study of 1 case showed molecular clustering with papillary RCC (158). In one study, tubulocystic RCC was commonly associated with papillary RCC, with both tumors showing gains in chromosomes 7 and 17 and loss of the Y chromosome (160). Furthermore, sometimes, separate tubulocystic RCC and papillary RCC tumors can be found in the same kidney, and at other times, they are closely admixed (163,164). These observations between tubulocystic RCC and papillary RCC, along with the overlapping immunohistochemical and molecular patterns, suggest a close relationship and raise the possibility that some tubulocystic RCCs could be a variant of papillary RCC. Some authors argued that tubulocystic RCC is a subset of papillary RCC, just as multiloculated clear cell RCC is considered to be a variant of clear cell RCC (111,160). More studies are needed to confirm the genetic relationship between the two types of tumor.
Hybrid oncocytic/chromophobe tumors (HOCT)
Hybrid oncocytic/chromophobe tumor (HOCT) is a rare tumor of the kidney, which is defined as a tumor having a mixture of cells with the morphological features of those seen in chromophobe RCC and renal oncocytoma. HOCTs have three clinicopathologic types: (I) sporadic; (II) those in association with renal oncocytosis/oncocytomatosis; and (III) those seen in patients with Birt-Hogg-Dubé syndrome (BHD) (165).
HOCT occur in adults without a sex predilection. There are no specific clinical signs and/or symptoms in patients with sporadic or HOCT associated with oncocytosis/oncocytomatosis. By contrast, HOCT in patients with BHD can present with a spontaneous pneumothorax associated with pulmonary cysts, and patients most often have multiple facial fibrofolliculomas or trichodiscoma (166-168). HOCTs (all three groups) seem to behave indolently, as no evidence of aggressive behavior has been documented (166-171).
From the data published to date, it appears that tumors from all three groups share similar morphological features—tumors were always composed of neoplastic cells with abundant granular eosinophilic “oncocytic” cytoplasm. An admixture of typical renal oncocytoma and chromophobe RCC areas is usually observed. Sometimes, the two areas were separated from each other (Figure 10), while at other times they mixed closely; i.e., scattered chromophobe cells in the background of a typical renal oncocytoma. The tumors were usually arranged in a solid-alveolar pattern. Some neoplastic cells can have a perinuclear halo with the presence of occasionally binucleate cells. These nuclear changes should only be found focally, and no raisinoid nuclei (as seen in classic chromophobe RCC) should be present (168-170,172,173). The immunohistochemical profiles of HOCTs in the three different groups differ slightly. All tumors tested were positive for cytokeratins AE1/AE3, EMA, and antimitochondrial antigen (AMA). CD117 was invariably positive in all three groups. Most tumors were positive for CK7, E-cadherin, and parvalbumin, whereas there was a lack of reactivity for racemase, CK20, CD10 and CA-IX. The tumors were frequently focally positive for VIM (165,169,171).
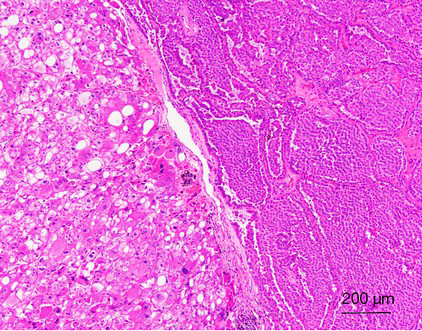
HOCT show significant molecular genetic heterogeneity. A useful and important diagnostic molecular genetic feature of HOCTs in patients with BHD is the presence of FLCN gene mutations, which are absent in the other groups. HOCTs in BHD also display multiple abnormalities that affect chromosomes 2, 3, 4, 5, 6, 13 and 18 (167,168). Sporadic HOCTs are characterized by multiple numerical aberrations (both mono- and polysomes), affecting chromosomes 1, 2, 6, 9, 10, 13, 17, 20, 21, and 22. No mutation in the VHL gene, KIT, or PDGFRA and no loss of heterozygosity for the small arm of chromosome 3 (3p) was detected (171). HOCTs associated with oncocytosis/oncocytomatosis usually show no chromosomal losses of chromosomes 1, 2, 6, 10, or 17. However, losses of chromosomes 1, 14, 21 and Y have been documented (173).
Acknowledgements
Funding: Supported by National Natural Science Foundation of China (81472391; 81101933; Q Rao), National Natural Science Foundation of China (81372743; XJ Zhou).
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Tickoo SK, dePeralta-Venturina MN, Harik LR, et al. Spectrum of epithelial neoplasms in end-stage renal disease: an experience from 66 tumor-bearing kidneys with emphasis on histologic patterns distinct from those in sporadic adult renal neoplasia. Am J Surg Pathol 2006;30:141-53. [PubMed]
- Gobbo S, Eble JN, Grignon DJ, et al. Clear cell papillary renal cell carcinoma: a distinct histopathologic and molecular genetic entity. Am J Surg Pathol 2008;32:1239-45. [PubMed]
- Aydin H, Chen L, Cheng L, et al. Clear cell tubulopapillary renal cell carcinoma: a study of 36 distinctive low-grade epithelial tumors of the kidney. Am J Surg Pathol 2010;34:1608-21. [PubMed]
- Adam J, Couturier J, Molinie V, et al. Clear-cell papillary renal cell carcinoma: 24 cases of a distinct low-grade renal tumour and a comparative genomic hybridization array study of seven cases. Histopathology 2011;58:1064-71. [PubMed]
- Rohan SM, Xiao Y, Liang Y, et al. Clear-cell papillary renal cell carcinoma: molecular and immunohistochemical analysis with emphasis on the von Hippel-Lindau gene and hypoxia-inducible factor pathway-related proteins. Mod Pathol 2011;24:1207-20. [PubMed]
- Williamson SR, Eble JN, Cheng L, et al. Clear cell papillary renal cell carcinoma: differential diagnosis and extended immunohistochemical profile. Mod Pathol 2013;26:697-708. [PubMed]
- Shi SS, Shen Q, Xia QY, et al. Clear cell papillary renal cell carcinoma: a clinicopathological study emphasizing ultrastructural features and cytogenetic heterogeneity. Int J Clin Exp Pathol 2013;6:2936-42. [PubMed]
- Williamson SR, Gupta NS, Eble JN, et al. Clear Cell Renal Cell Carcinoma With Borderline Features of Clear Cell Papillary Renal Cell Carcinoma: Combined Morphologic, Immunohistochemical, and Cytogenetic Analysis. Am J Surg Pathol 2015;39:1502-10. [PubMed]
- Aron M, Chang E, Herrera L, et al. Clear cell-papillary renal cell carcinoma of the kidney not associated with end-stage renal disease: clinicopathologic correlation with expanded immunophenotypic and molecular characterization of a large cohort with emphasis on relationship with renal angiomyoadenomatous tumor. Am J Surg Pathol 2015;39:873-88. [PubMed]
- Deml KF, Schildhaus HU, Comperat E, et al. Clear cell papillary renal cell carcinoma and renal angiomyoadenomatous tumor: two variants of a morphologic, immunohistochemical, and genetic distinct entity of renal cell carcinoma. Am J Surg Pathol 2015;39:889-901. [PubMed]
- Michal M, Hes O, Havlicek F. Benign renal angiomyoadenomatous tumor: a previously unreported renal tumor. Ann Diagn Pathol 2000;4:311-5. [PubMed]
- Williamson SR, Zhang S, Eble JN, et al. Clear cell papillary renal cell carcinoma-like tumors in patients with von Hippel-Lindau disease are unrelated to sporadic clear cell papillary renal cell carcinoma. Am J Surg Pathol 2013;37:1131-9. [PubMed]
- Zhou H, Zheng S, Truong LD, et al. Clear cell papillary renal cell carcinoma is the fourth most common histologic type of renal cell carcinoma in 290 consecutive nephrectomies for renal cell carcinoma. Hum Pathol 2014;45:59-64. [PubMed]
- Rao P, Monzon F, Jonasch E, et al. Clear cell papillary renal cell carcinoma in patients with von Hippel-Lindau syndrome--clinicopathological features and comparative genomic analysis of 3 cases. Hum Pathol 2014;45:1966-72. [PubMed]
- Srigley JR, Delahunt B, Eble JN, et al. The International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia. Am J Surg Pathol 2013;37:1469-89. [PubMed]
- Leroy X, Camparo P, Gnemmi V, et al. Clear cell papillary renal cell carcinoma is an indolent and low-grade neoplasm with overexpression of cyclin-D1. Histopathology 2014;64:1032-6. [PubMed]
- Munari E, Marchionni L, Chitre A, et al. Clear cell papillary renal cell carcinoma: micro-RNA expression profiling and comparison with clear cell renal cell carcinoma and papillary renal cell carcinoma. Hum Pathol 2014;45:1130-8. [PubMed]
- Slaby O, Redova M, Poprach A, et al. Identification of MicroRNAs associated with early relapse after nephrectomy in renal cell carcinoma patients. Genes Chromosomes Cancer 2012;51:707-16. [PubMed]
- Chan SY, Loscalzo J. MicroRNA-210: a unique and pleiotropic hypoxamir. Cell Cycle 2010;9:1072-83. [PubMed]
- Chang W, Lee CY, Park JH, et al. Survival of hypoxic human mesenchymal stem cells is enhanced by a positive feedback loop involving miR-210 and hypoxia-inducible factor 1. J Vet Sci 2013;14:69-76. [PubMed]
- Lawrie CH, Larrea E, Larrinaga G, et al. Targeted next-generation sequencing and non-coding RNA expression analysis of clear cell papillary renal cell carcinoma suggests distinct pathological mechanisms from other renal tumour subtypes. J Pathol 2014;232:32-42. [PubMed]
- Fisher KE, Yin-Goen Q, Alexis D, et al. Gene expression profiling of clear cell papillary renal cell carcinoma: comparison with clear cell renal cell carcinoma and papillary renal cell carcinoma. Mod Pathol 2014;27:222-30. [PubMed]
- Wolfe A, Dobin SM, Grossmann P, et al. Clonal trisomies 7,10 and 12, normal 3p and absence of VHL gene mutation in a clear cell tubulopapillary carcinoma of the kidney. Virchows Arch 2011;459:457-63. [PubMed]
- Kuroda N, Shiotsu T, Kawada C, et al. Clear cell papillary renal cell carcinoma and clear cell renal cell carcinoma arising in acquired cystic disease of the kidney: an immunohistochemical and genetic study. Ann Diagn Pathol 2011;15:282-5. [PubMed]
- Inoue T, Matsuura K, Yoshimoto T, et al. Genomic profiling of renal cell carcinoma in patients with end-stage renal disease. Cancer Sci 2012;103:569-76. [PubMed]
- Rao Q, Cheng L, Xia QY, et al. Cathepsin K expression in a wide spectrum of perivascular epithelioid cell neoplasms (PEComas): a clinicopathological study emphasizing extrarenal PEComas. Histopathology 2013;62:642-50. [PubMed]
- Argani P, Aulmann S, Illei PB, et al. A distinctive subset of PEComas harbors TFE3 gene fusions. Am J Surg Pathol 2010;34:1395-406. [PubMed]
- Argani P, Olgac S, Tickoo SK, et al. Xp11 translocation renal cell carcinoma in adults: expanded clinical, pathologic, and genetic spectrum. Am J Surg Pathol 2007;31:1149-60. [PubMed]
- Malouf GG, Su X, Yao H, et al. Next-generation sequencing of translocation renal cell carcinoma reveals novel RNA splicing partners and frequent mutations of chromatin-remodeling genes. Clin Cancer Res 2014;20:4129-40. [PubMed]
- Tsuda M, Davis IJ, Argani P, et al. TFE3 fusions activate MET signaling by transcriptional up-regulation, defining another class of tumors as candidates for therapeutic MET inhibition. Cancer Res 2007;67:919-29. [PubMed]
- Medendorp K, van Groningen JJ, Schepens M, et al. Molecular mechanisms underlying the MiT translocation subgroup of renal cell carcinomas. Cytogenet Genome Res 2007;118:157-65. [PubMed]
- Argani P, Ladanyi M. Translocation carcinomas of the kidney. Clin Lab Med 2005;25:363-78. [PubMed]
- Delahunt B, Egevad L, Montironi R, et al. International Society of Urological Pathology (ISUP) consensus conference on renal neoplasia: rationale and organization. Am J Surg Pathol 2013;37:1463-8. [PubMed]
- Komai Y, Fujiwara M, Fujii Y, et al. Adult Xp11 translocation renal cell carcinoma diagnosed by cytogenetics and immunohistochemistry. Clin Cancer Res 2009;15:1170-6. [PubMed]
- Argani P, Lae M, Ballard ET, et al. Translocation carcinomas of the kidney after chemotherapy in childhood. J Clin Oncol 2006;24:1529-34. [PubMed]
- Armah HB, Parwani AV, Surti U, et al. Xp11.2 translocation renal cell carcinoma occurring during pregnancy with a novel translocation involving chromosome 19: a case report with review of the literature. Diagn Pathol 2009;4:15. [PubMed]
- Nouh MA, Kuroda N, Yamashita M, et al. Renal cell carcinoma in patients with end-stage renal disease: relationship between histological type and duration of dialysis. BJU Int 2010;105:620-7. [PubMed]
- Geller JI, Argani P, Adeniran A, et al. Translocation renal cell carcinoma: lack of negative impact due to lymph node spread. Cancer 2008;112:1607-16. [PubMed]
- Koie T, Yoneyama T, Hashimoto Y, et al. An aggressive course of Xp11 translocation renal cell carcinoma in a 28-year-old man. Int J Urol 2009;16:333-5. [PubMed]
- Rao Qiu, Guan Bing, Zhou XJ. Xp11.2 Translocation renal cell carcinomas have a poorer prognosis than non-Xp11.2 translocation carcinomas in children and young adults: a meta-analysis. Int J Surg Pathol 2010;18:458-64. [PubMed]
- Rao Q, Chen JY, Wang JD, et al. Renal cell carcinoma in children and young adults: clinicopathological, immunohistochemical, and VHL gene analysis of 46 cases with follow-up. Int J Surg Pathol 2011;19:170-9. [PubMed]
- Rais-Bahrami S, Drabick JJ, De Marzo AM, et al. Xp11 translocation renal cell carcinoma: delayed but massive and lethal metastases of a chemotherapy-associated secondary malignancy. Urology 2007;70:178.e3-6.
- Armah HB, Parwani AV. Xp11.2 translocation renal cell carcinoma. Arch Pathol Lab Med 2010;134:124-9. [PubMed]
- Choueiri TK, Lim ZD, Hirsch MS, et al. Vascular endothelial growth factor-targeted therapy for the treatment of adult metastatic Xp11.2 translocation renal cell carcinoma. Cancer 2010;116:5219-25. [PubMed]
- Malouf GG, Camparo P, Oudard S, et al. Targeted agents in metastatic Xp11 translocation/TFE3 gene fusion renal cell carcinoma (RCC): a report from the Juvenile RCC Network. Ann Oncol 2010;21:1834-8. [PubMed]
- Rao Q, Williamson SR, Zhang S, et al. TFE3 break-apart FISH has a higher sensitivity for Xp11.2 translocation-associated renal cell carcinoma compared with TFE3 or cathepsin K immunohistochemical staining alone: expanding the morphologic spectrum. Am J Surg Pathol 2013;37:804-15. [PubMed]
- Green WM, Yonescu R, Morsberger L, et al. Utilization of a TFE3 break-apart FISH assay in a renal tumor consultation service. Am J Surg Pathol 2013;37:1150-63. [PubMed]
- Martignoni G, Pea M, Gobbo S, et al. Cathepsin-K immunoreactivity distinguishes MiTF/TFE family renal translocation carcinomas from other renal carcinomas. Mod Pathol 2009;22:1016-22. [PubMed]
- Martignoni G, Gobbo S, Camparo P, et al. Differential expression of cathepsin K in neoplasms harboring TFE3 gene fusions. Mod Pathol 2011;24:1313-9. [PubMed]
- Camparo P, Vasiliu V, Molinie V, et al. Renal translocation carcinomas: clinicopathologic, immunohistochemical, and gene expression profiling analysis of 31 cases with a review of the literature. Am J Surg Pathol 2008;32:656-70. [PubMed]
- Mosquera JM, Dal Cin P, Mertz KD, et al. Validation of a TFE3 break-apart FISH assay for Xp11.2 translocation renal cell carcinomas. Diagn Mol Pathol 2011;20:129-37. [PubMed]
- Kim SH, Choi Y, Jeong HY, et al. Usefulness of a break-apart FISH assay in the diagnosis of Xp11.2 translocation renal cell carcinoma. Virchows Arch 2011;459:299-306. [PubMed]
- Klatte T, Streubel B, Wrba F, et al. Renal cell carcinoma associated with transcription factor E3 expression and Xp11.2 translocation: incidence, characteristics, and prognosis. Am J Clin Pathol 2012;137:761-8. [PubMed]
- Rao Q, Shen Q, Xia QY, et al. PSF/SFPQ is a very common gene fusion partner in TFE3 rearrangement-associated perivascular epithelioid cell tumors (PEComas) and melanotic Xp11 translocation renal cancers: clinicopathologic, immunohistochemical, and molecular characteristics suggesting classification as a distinct entity. Am J Surg Pathol 2015;39:1181-96. [PubMed]
- Chang IW, Huang HY, Sung MT. Melanotic Xp11 translocation renal cancer: a case with PSF-TFE3 gene fusion and up-regulation of melanogenetic transcripts. Am J Surg Pathol 2009;33:1894-901. [PubMed]
- Tanaka M, Kato K, Gomi K, et al. Perivascular epithelioid cell tumor with SFPQ/PSF-TFE3 gene fusion in a patient with advanced neuroblastoma. Am J Surg Pathol 2009;33:1416-20. [PubMed]
- LeGallo RD, Stelow EB, Sukov WR, et al. Melanotic xp11.2 neoplasm of the ovary: report of a unique case. Am J Surg Pathol 2012;36:1410-4. [PubMed]
- Argani P, Hawkins A, Griffin CA, et al. A distinctive pediatric renal neoplasm characterized by epithelioid morphology, basement membrane production, focal HMB45 immunoreactivity, and t(6;11)(p21.1;q12) chromosome translocation. Am J Pathol 2001;158:2089-96. [PubMed]
- Davis IJ, Hsi BL, Arroyo JD, et al. Cloning of an Alpha-TFEB fusion in renal tumors harboring the t(6;11)(p21;q13) chromosome translocation. Proc Natl Acad Sci U S A 2003;100:6051-6. [PubMed]
- Kuiper RP, Schepens M, Thijssen J, et al. Upregulation of the transcription factor TFEB in t(6;11)(p21;q13)-positive renal cell carcinomas due to promoter substitution. Hum Mol Genet 2003;12:1661-9. [PubMed]
- Argani P, Lae M, Hutchinson B, et al. Renal carcinomas with the t(6;11)(p21;q12): clinicopathologic features and demonstration of the specific alpha-TFEB gene fusion by immunohistochemistry, RT-PCR, and DNA PCR. Am J Surg Pathol 2005;29:230-40. [PubMed]
- Pecciarini L, Cangi MG, Lo Cunsolo C, et al. Characterization of t(6;11)(p21;q12) in a renal-cell carcinoma of an adult patient. Genes Chromosomes Cancer 2007;46:419-26. [PubMed]
- Hora M, Hes O, Urge T, et al. A distinctive translocation carcinoma of the kidney Int Urol Nephrol 2009;41:553-7. ["rosette-like forming," t(6;11), HMB45-positive renal tumor]. [PubMed]
- Zhan HQ, Wang CF, Zhu XZ, et al. Renal cell carcinoma with t(6;11) translocation: a patient case with a novel Alpha-TFEB fusion point. J Clin Oncol 2010;28:e709-13. [PubMed]
- Suárez-Vilela D, Izquierdo-García F, Méndez-Álvarez JR, et al. Renal translocation carcinoma with expression of TFEB: presentation of a case with distinctive histological and immunohistochemical features. Int J Surg Pathol 2011;19:506-9. [PubMed]
- Inamura K, Fujiwara M, Togashi Y, et al. Diverse fusion patterns and heterogeneous clinicopathologic features of renal cell carcinoma with t(6;11) translocation. Am J Surg Pathol 2012;36:35-42. [PubMed]
- Petersson F, Vanecek T, Michal M, et al. A distinctive translocation carcinoma of the kidney; "rosette forming," t(6;11), HMB45-positive renal tumor: a histomorphologic, immunohistochemical, ultrastructural, and molecular genetic study of 4 cases. Hum Pathol 2012;43:726-36. [PubMed]
- Rao Q, Liu B, Cheng L, et al. Renal cell carcinomas with t(6;11)(p21;q12): A clinicopathologic study emphasizing unusual morphology, novel alpha-TFEB gene fusion point, immunobiomarkers, and ultrastructural features, as well as detection of the gene fusion by fluorescence in situ hybridization. Am J Surg Pathol 2012;36:1327-38. [PubMed]
- Argani P, Yonescu R, Morsberger L, et al. Molecular confirmation of t(6;11)(p21;q12) renal cell carcinoma in archival paraffin-embedded material using a break-apart TFEB FISH assay expands its clinicopathologic spectrum. Am J Surg Pathol 2012;36:1516-26. [PubMed]
- Rao Q, Zhang XM, Tu P, et al. Renal cell carcinomas with t(6;11)(p21;q12) presenting with tubulocystic renal cell carcinoma-like features. Int J Clin Exp Pathol 2013;6:1452-7. [PubMed]
- Smith NE, Illei PB, Allaf M, et al. t(6;11) renal cell carcinoma (RCC): expanded immunohistochemical profile emphasizing novel RCC markers and report of 10 new genetically confirmed cases. Am J Surg Pathol 2014;38:604-14. [PubMed]
- Barletta JA, Hornick JL. Succinate dehydrogenase-deficient tumors: diagnostic advances and clinical implications. Adv Anat Pathol 2012;19:193-203. [PubMed]
- Gill AJ. Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology 2012;44:285-92. [PubMed]
- Doyle LA, Hornick JL. Gastrointestinal stromal tumours: from KIT to succinate dehydrogenase. Histopathology 2014;64:53-67. [PubMed]
- Ricketts CJ, Shuch B, Vocke CD, et al. Succinate dehydrogenase kidney cancer: an aggressive example of the Warburg effect in cancer. J Urol 2012;188:2063-71. [PubMed]
- Vanharanta S, Buchta M, McWhinney SR, et al. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet 2004;74:153-9. [PubMed]
- Housley SL, Lindsay RS, Young B, et al. Renal carcinoma with giant mitochondria associated with germ-line mutation and somatic loss of the succinate dehydrogenase B gene. Histopathology 2010;56:405-8. [PubMed]
- Gill AJ, Pachter NS, Chou A, et al. Renal tumors associated with germline SDHB mutation show distinctive morphology. Am J Surg Pathol 2011;35:1578-85. [PubMed]
- Miettinen M, Sarlomo-Rikala M, McCue P, et al. Mapping of succinate dehydrogenase losses in 2258 epithelial neoplasms. Appl Immunohistochem Mol Morphol 2014;22:31-6. [PubMed]
- Gill AJ, Hes O, Papathomas T, et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: a morphologically distinct entity: a clinicopathologic series of 36 tumors from 27 patients. Am J Surg Pathol 2014;38:1588-602. [PubMed]
- Williamson SR, Eble JN, Amin MB, et al. Succinate dehydrogenase-deficient renal cell carcinoma: detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod Pathol 2015;28:80-94. [PubMed]
- Papathomas TG, Gaal J, Corssmit EP, et al. Non-pheochromocytoma (PCC)/paraganglioma (PGL) tumors in patients with succinate dehydrogenase-related PCC-PGL syndromes: a clinicopathological and molecular analysis. Eur J Endocrinol 2014;170:1-12. [PubMed]
- Yakirevich E, Ali SM, Mega A, et al. A Novel SDHA-deficient Renal Cell Carcinoma Revealed by Comprehensive Genomic Profiling. Am J Surg Pathol 2015;39:858-63. [PubMed]
- Haller F, Moskalev EA, Faucz FR, et al. Aberrant DNA hypermethylation of SDHC: a novel mechanism of tumor development in Carney triad. Endocr Relat Cancer 2014;21:567-77. [PubMed]
- Killian JK, Miettinen M, Walker RL, et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci Transl Med 2014;6:268ra177.
- Burnichon N, Briere JJ, Libe R, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet 2010;19:3011-20. [PubMed]
- Fleming S, Mayer NJ, Vlatkovic LJ, et al. Signalling pathways in succinate dehydrogenase B-associated renal carcinoma. Histopathology 2014;64:477-83. [PubMed]
- Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 1994;263:1281-4. [PubMed]
- Iwahara T, Fujimoto J, Wen D, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997;14:439-49. [PubMed]
- Lawrence B, Perez-Atayde A, Hibbard MK, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol 2000;157:377-84. [PubMed]
- Ma Z, Cools J, Marynen P, et al. Inv(2)(p23q35) in anaplastic large-cell lymphoma induces constitutive anaplastic lymphoma kinase (ALK) tyrosine kinase activation by fusion to ATIC, an enzyme involved in purine nucleotide biosynthesis. Blood 2000;95:2144-9. [PubMed]
- Bridge JA, Kanamori M, Ma Z, et al. Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory myofibroblastic tumor. Am J Pathol 2001;159:411-5. [PubMed]
- Dirks WG, Fahnrich S, Lis Y, et al. Expression and functional analysis of the anaplastic lymphoma kinase (ALK) gene in tumor cell lines. Int J Cancer 2002;100:49-56. [PubMed]
- Gascoyne RD, Lamant L, Martin-Subero JI, et al. ALK-positive diffuse large B-cell lymphoma is associated with Clathrin-ALK rearrangements: report of 6 cases. Blood 2003;102:2568-73. [PubMed]
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561-6. [PubMed]
- Lin E, Li L, Guan Y, et al. Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancers. Mol Cancer Res 2009;7:1466-76. [PubMed]
- Debelenko LV, Raimondi SC, Daw N, et al. Renal cell carcinoma with novel VCL-ALK fusion: new representative of ALK-associated tumor spectrum. Mod Pathol 2011;24:430-42. [PubMed]
- Mariño-Enríquez A, Ou WB, Weldon CB, et al. ALK rearrangement in sickle cell trait-associated renal medullary carcinoma. Genes Chromosomes Cancer 2011;50:146-53. [PubMed]
- Sugawara E, Togashi Y, Kuroda N, et al. Identification of anaplastic lymphoma kinase fusions in renal cancer: large-scale immunohistochemical screening by the intercalated antibody-enhanced polymer method. Cancer 2012;118:4427-36. [PubMed]
- Sukov WR, Hodge JC, Lohse CM, et al. ALK alterations in adult renal cell carcinoma: frequency, clinicopathologic features and outcome in a large series of consecutively treated patients. Mod Pathol 2012;25:1516-25. [PubMed]
- Hodge JC, Pearce KE, Sukov WR. Distinct ALK-rearranged and VCL-negative papillary renal cell carcinoma variant in two adults without sickle cell trait. Mod Pathol 2013;26:604-5. [PubMed]
- Lee C, Park JW, Suh JH, et al. ALK-Positive Renal Cell Carcinoma in a Large Series of Consecutively Resected Korean Renal Cell Carcinoma Patients. Korean J Pathol 2013;47:452-7. [PubMed]
- Smith NE, Deyrup AT, Marino-Enriquez A, et al. VCL-ALK renal cell carcinoma in children with sickle-cell trait: the eighth sickle-cell nephropathy? Am J Surg Pathol 2014;38:858-63. [PubMed]
- Chiarle R, Voena C, Ambrogio C, et al. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer 2008;8:11-23. [PubMed]
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363:1693-703. [PubMed]
- Chihara D, Suzuki R. More on crizotinib. N Engl J Med 2011;364:776-7; author reply 8. [PubMed]
- Sangoi AR, Higgins JP. Bilateral mixed epithelial stromal tumor in an end-stage renal disease patient: the first case report. Hum Pathol 2008;39:142-6. [PubMed]
- Hamamoto S, Okamura T, Mizuno K, et al. Renal oncocytoma with bilateral synchronous renal cell carcinoma in a patient undergoing long-term hemodialysis. Int J Urol 2008;15:87-9. [PubMed]
- Sakura M, Masuda H, Saito K, et al. Collecting duct carcinoma with acquired cystic disease of the kidney in a long-term hemodialysis patient. Int J Urol 2008;15:93-5. [PubMed]
- Nishihara K, Shomori K, Nagashima Y, et al. Chromophobe renal cell carcinoma and 'capsulomas' with acquired cystic disease of the kidney in a long-term hemodialysis patient. Int J Urol 2008;15:543-5. [PubMed]
- Srigley JR, Delahunt B. Uncommon and recently described renal carcinomas. Mod Pathol 2009;22 Suppl 2:S2-S23. [PubMed]
- Kuroda N, Tamura M, Hamaguchi N, et al. Acquired cystic disease-associated renal cell carcinoma with sarcomatoid change and rhabdoid features. Ann Diagn Pathol 2011;15:462-6. [PubMed]
- Sule N, Yakupoglu U, Shen SS, et al. Calcium oxalate deposition in renal cell carcinoma associated with acquired cystic kidney disease: a comprehensive study. Am J Surg Pathol 2005;29:443-51. [PubMed]
- Cossu-Rocca P, Eble JN, Zhang S, et al. Acquired cystic disease-associated renal tumors: an immunohistochemical and fluorescence in situ hybridization study. Mod Pathol 2006;19:780-7. [PubMed]
- Pan CC, Chen YJ, Chang LC, et al. Immunohistochemical and molecular genetic profiling of acquired cystic disease-associated renal cell carcinoma. Histopathology 2009;55:145-53. [PubMed]
- Enoki Y, Katoh G, Okabe H, et al. Clinicopathological features and CD57 expression in renal cell carcinoma in acquired cystic disease of the kidneys: with special emphasis on a relation to the duration of haemodialysis, the degree of calcium oxalate deposition, histological type, and possible tumorigenesis. Histopathology 2010;56:384-94. [PubMed]
- Kuroda N, Yamashita M, Kakehi Y, et al. Acquired cystic disease-associated renal cell carcinoma: an immunohistochemical and fluorescence in situ hybridization study. Med Mol Morphol 2011;44:228-32. [PubMed]
- Bhatnagar R, Alexiev BA. Renal-cell carcinomas in end-stage kidneys: a clinicopathological study with emphasis on clear-cell papillary renal-cell carcinoma and acquired cystic kidney disease-associated carcinoma. Int J Surg Pathol 2012;20:19-28. [PubMed]
- Ahn S, Kwon GY, Cho YM, et al. Acquired cystic disease-associated renal cell carcinoma: further characterization of the morphologic and immunopathologic features. Med Mol Morphol 2013;46:225-32. [PubMed]
- Kuroda N, Shiotsu T, Hes O, et al. Acquired cystic disease-associated renal cell carcinoma with gain of chromosomes 3, 7, and 16, gain of chromosome X, and loss of chromosome Y. Med Mol Morphol 2010;43:231-4. [PubMed]
- Kuntz E, Yusenko MV, Nagy A, et al. Oligoarray comparative genomic hybridization of renal cell tumors that developed in patients with acquired cystic renal disease. Hum Pathol 2010;41:1345-9. [PubMed]
- Tajima S, Waki M, Doi W, et al. Acquired cystic disease-associated renal cell carcinoma with a focal sarcomatoid component: Report of a case showing more pronounced polysomy of chromosomes 3 and 16 in the sarcomatoid component. Pathol Int 2015;65:89-94. [PubMed]
- Kiuru M, Launonen V, Hietala M, et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol 2001;159:825-9. [PubMed]
- Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 2002;30:406-10. [PubMed]
- Barrisford GW, Singer EA, Rosner IL, et al. Familial renal cancer: molecular genetics and surgical management. Int J Surg Oncol 2011;2011:658767.
- Lehtonen HJ. Hereditary leiomyomatosis and renal cell cancer: update on clinical and molecular characteristics. Fam Cancer 2011;10:397-411. [PubMed]
- Grubb RL, 3rd, Franks ME, Toro J, et al. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol 2007;177:2074-9; discussion 9-80. [PubMed]
- Sanz-Ortega J, Vocke C, Stratton P, et al. Morphologic and molecular characteristics of uterine leiomyomas in hereditary leiomyomatosis and renal cancer (HLRCC) syndrome. Am J Surg Pathol 2013;37:74-80. [PubMed]
- Merino MJ, Torres-Cabala C, Pinto P, et al. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol 2007;31:1578-85. [PubMed]
- Udager AM, Alva A, Chen YB, et al. Hereditary leiomyomatosis and renal cell carcinoma (HLRCC): a rapid autopsy report of metastatic renal cell carcinoma. Am J Surg Pathol 2014;38:567-77. [PubMed]
- Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 2003;73:95-106. [PubMed]
- Alam NA, Rowan AJ, Wortham NC, et al. Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet 2003;12:1241-52. [PubMed]
- Gardie B, Remenieras A, Kattygnarath D, et al. Novel FH mutations in families with hereditary leiomyomatosis and renal cell cancer (HLRCC) and patients with isolated type 2 papillary renal cell carcinoma. J Med Genet 2011;48:226-34. [PubMed]
- Isaacs JS, Jung YJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 2005;8:143-53. [PubMed]
- Alderson NL, Wang Y, Blatnik M, et al. S-(2-Succinyl)cysteine: a novel chemical modification of tissue proteins by a Krebs cycle intermediate. Arch Biochem Biophys 2006;450:1-8. [PubMed]
- Nagai R, Brock JW, Blatnik M, et al. Succination of protein thiols during adipocyte maturation: a biomarker of mitochondrial stress. J Biol Chem 2007;282:34219-28. [PubMed]
- Frizzell N, Rajesh M, Jepson MJ, et al. Succination of thiol groups in adipose tissue proteins in diabetes: succination inhibits polymerization and secretion of adiponectin. J Biol Chem 2009;284:25772-81. [PubMed]
- Joseph NM, Solomon DA, Frizzell N, et al. Morphology and Immunohistochemistry for 2SC and FH Aid in Detection of Fumarate Hydratase Gene Aberrations in Uterine Leiomyomas From Young Patients. Am J Surg Pathol 2015;39:1529-39. [PubMed]
- Bardella C, El-Bahrawy M, Frizzell N, et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol 2011;225:4-11. [PubMed]
- Chen YB, Brannon AR, Toubaji A, et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol 2014;38:627-37. [PubMed]
- Jung SJ, Chung JI, Park SH, et al. Thyroid follicular carcinoma-like tumor of kidney: a case report with morphologic, immunohistochemical, and genetic analysis. Am J Surg Pathol 2006;30:411-5. [PubMed]
- Sterlacci W, Verdorfer I, Gabriel M, et al. Thyroid follicular carcinoma-like renal tumor: a case report with morphologic, immunophenotypic, cytogenetic, and scintigraphic studies. Virchows Arch 2008;452:91-5. [PubMed]
- Amin MB, Gupta R, Ondrej H, et al. Primary thyroid-like follicular carcinoma of the kidney: report of 6 cases of a histologically distinctive adult renal epithelial neoplasm. Am J Surg Pathol 2009;33:393-400. [PubMed]
- Dhillon J, Tannir NM, Matin SF, et al. Thyroid-like follicular carcinoma of the kidney with metastases to the lungs and retroperitoneal lymph nodes. Hum Pathol 2011;42:146-50. [PubMed]
- Alessandrini L, Fassan M, Gardiman MP, et al. Thyroid-like follicular carcinoma of the kidney: report of two cases with detailed immunohistochemical profile and literature review. Virchows Arch 2012;461:345-50. [PubMed]
- Khoja HA, Almutawa A, Binmahfooz A, et al. Papillary thyroid carcinoma-like tumor of the kidney: a case report. Int J Surg Pathol 2012;20:411-5. [PubMed]
- Malde S, Sheikh I, Woodman I, et al. Primary thyroid-like follicular renal cell carcinoma: an emerging entity. Case Rep Pathol 2013;2013:687427.
- Chougule A, Bal A, Das A, et al. Thyroid-like follicular renal cell carcinoma: an emerging morphological variant. Pathology 2014;46:657-60. [PubMed]
- Dhillon J, Mohanty SK, Krishnamurthy S. Cytologic diagnosis of thyroid-like follicular carcinoma of the kidney: a case report. Diagn Cytopathol 2014;42:273-7. [PubMed]
- Ghaouti M, Roquet L, Baron M, et al. Thyroid-like follicular carcinoma of the kidney: a case report and review of the literature. Diagn Pathol 2014;9:186. [PubMed]
- Lin YZ, Wei Y, Xu N, et al. Thyroid-like follicular carcinoma of the kidney: A report of two cases and literature review. Oncol Lett 2014;7:1796-802. [PubMed]
- Vicens RA, Balachandran A, Guo CC, et al. Multimodality imaging of thyroid-like follicular renal cell carcinoma with lung metastases, a new emerging tumor entity. Abdom Imaging 2014;39:388-93. [PubMed]
- Wu WW, Chu JT, Nael A, et al. Thyroid-like follicular carcinoma of the kidney in a young patient with history of pediatric acute lymphoblastic leukemia. Case Rep Pathol 2014;2014:313974.
- Ohe C, Kuroda N, Pan CC, et al. A unique renal cell carcinoma with features of papillary renal cell carcinoma and thyroid-like carcinoma: a morphological, immunohistochemical and genetic study. Histopathology 2010;57:494-7. [PubMed]
- Graux C, Cools J, Michaux L, et al. Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: from thymocyte to lymphoblast. Leukemia 2006;20:1496-510. [PubMed]
- MacLennan GT, Farrow GM, Bostwick DG. Low-grade collecting duct carcinoma of the kidney: report of 13 cases of low-grade mucinous tubulocystic renal carcinoma of possible collecting duct origin. Urology 1997;50:679-84. [PubMed]
- Azoulay S, Vieillefond A, Paraf F, et al. Tubulocystic carcinoma of the kidney: a new entity among renal tumors. Virchows Arch 2007;451:905-9. [PubMed]
- Yang XJ, Zhou M, Hes O, et al. Tubulocystic carcinoma of the kidney: clinicopathologic and molecular characterization. Am J Surg Pathol 2008;32:177-87. [PubMed]
- Amin MB, MacLennan GT, Gupta R, et al. Tubulocystic carcinoma of the kidney: clinicopathologic analysis of 31 cases of a distinctive rare subtype of renal cell carcinoma. Am J Surg Pathol 2009;33:384-92. [PubMed]
- Zhou M, Yang XJ, Lopez JI, et al. Renal tubulocystic carcinoma is closely related to papillary renal cell carcinoma: implications for pathologic classification. Am J Surg Pathol 2009;33:1840-9. [PubMed]
- Tran T, Jones CL, Williamson SR, et al. Tubulocystic renal cell carcinoma is an entity that is immunohistochemically and genetically distinct from papillary renal cell carcinoma. Histopathology 2015. [Epub ahead of print]. [PubMed]
- Osunkoya AO, Young AN, Wang W, et al. Comparison of gene expression profiles in tubulocystic carcinoma and collecting duct carcinoma of the kidney. Am J Surg Pathol 2009;33:1103-6. [PubMed]
- Deshmukh M, Shet T, Bakshi G, et al. Tubulocystic carcinoma of kidney associated with papillary renal cell carcinoma. Indian J Pathol Microbiol 2011;54:127-30. [PubMed]
- Chen N, Nie L, Gong J, et al. Gains of chromosomes 7 and 17 in tubulocystic carcinoma of kidney: two cases with fluorescence in situ hybridisation analysis. J Clin Pathol 2014;67:1006-9. [PubMed]
- Hes O, Petersson F, Kuroda N, et al. Renal hybrid oncocytic/chromophobe tumors - a review. Histol Histopathol 2013;28:1257-64. [PubMed]
- Abbosh PH, Grubb RL 3rd, Cao D, et al. Hybrid renal tumors in Birt-Hogg-Dube syndrome. J Urol 2011;186:2413-4. [PubMed]
- Adley BP, Smith ND, Nayar R, et al. Birt-Hogg-Dube syndrome: clinicopathologic findings and genetic alterations. Arch Pathol Lab Med 2006;130:1865-70. [PubMed]
- Pavlovich CP, Walther MM, Eyler RA, et al. Renal tumors in the Birt-Hogg-Dube syndrome. Am J Surg Pathol 2002;26:1542-52. [PubMed]
- Mai KT, Dhamanaskar P, Belanger E, et al. Hybrid chromophobe renal cell neoplasm. Pathol Res Pract 2005;201:385-9. [PubMed]
- Nagashima Y, Furuya M, Gotohda H, et al. FLCN gene-mutated renal cell neoplasms: mother and daughter cases with a novel germline mutation. Int J Urol 2012;19:468-70. [PubMed]
- Petersson F, Gatalica Z, Grossmann P, et al. Sporadic hybrid oncocytic/chromophobe tumor of the kidney: a clinicopathologic, histomorphologic, immunohistochemical, ultrastructural, and molecular cytogenetic study of 14 cases. Virchows Arch 2010;456:355-65. [PubMed]
- Delongchamps NB, Galmiche L, Eiss D, et al. Hybrid tumour 'oncocytoma-chromophobe renal cell carcinoma' of the kidney: a report of seven sporadic cases. BJU Int 2009;103:1381-4. [PubMed]
- Gobbo S, Eble JN, Delahunt B, et al. Renal cell neoplasms of oncocytosis have distinct morphologic, immunohistochemical, and cytogenetic profiles. Am J Surg Pathol 2010;34:620-6. [PubMed]