
Overproduction of reactive oxygen species - obligatory or not for induction of apoptosis by anticancer drugs
Abstract
Many studies demonstrate that conventional anticancer drugs elevate intracellular level of reactive oxygen species (ROS) and alter redox-homeostasis of cancer cells. It is widely accepted that anticancer effect of these chemotherapeutics is due to induction of oxidative stress and ROS-mediated apoptosis in cancer. On the other hand, the harmful side effects of conventional anticancer chemotherapy are also due to increased production of ROS and disruption of redox-homeostasis of normal cells and tissues. This article describes the mechanisms for triggering and modulation of apoptosis through ROS-dependent and ROS-independent pathways. We try to answer the question: "Is it possible to induce highly specific apoptosis only in cancer cells, without overproduction of ROS, as well as without harmful effects on normal cells and tissues?" The review also suggests a new therapeutic strategy for selective killing of cancer cells, without significant impact on viability of normal cells and tissues, by combining anticancer drugs with redox-modulators, affecting specific signaling pathways and avoiding oxidative stress.
Keywords: Cancer; reactive oxygen species; chemotherapy; ROS-independent apoptosis; ROS-dependent apoptosis
Submitted Apr 26, 2016. Accepted for publication Jun 30, 2016.
doi: 10.21147/j.issn.1000-9604.2016.04.01
Reactive oxygen species as activator(s) of cellular redox-signaling and their impact for carcinogenesis
In the past three decades, a large number of studies indicate that reactive oxygen species (ROS) are generated by different xenobiotics, including anticancer drugs, and they also act as secondary messengers in cell signaling and are essential for various biological processes in normal cells (1-18). ROS (superoxide radical, hydroxyl radical, hydroperoxyl radical, hydrogen peroxide) are produced by: 1) mitochondrial electron-transport chain (8-11); 2) nicotinamide adenine dinucleotide phosphate (NADPH)-dependent oxidase (NOX) complex (10, 12); 3) enzymes such as cytochrome P450 (13, 14), xanthine oxidase (15, 16), as well as other intracellular sources (Figure 1). The level of physiologically generated ROS is controlled by enzymatic and non-enzymatic intracellular antioxidant systems, which are connected with different functional pathways (16-19). The cellular oxidative stress is a result of redox-imbalance due to enhancement of ROS or suppression and crash of antioxidant systems (16, 17, 20).
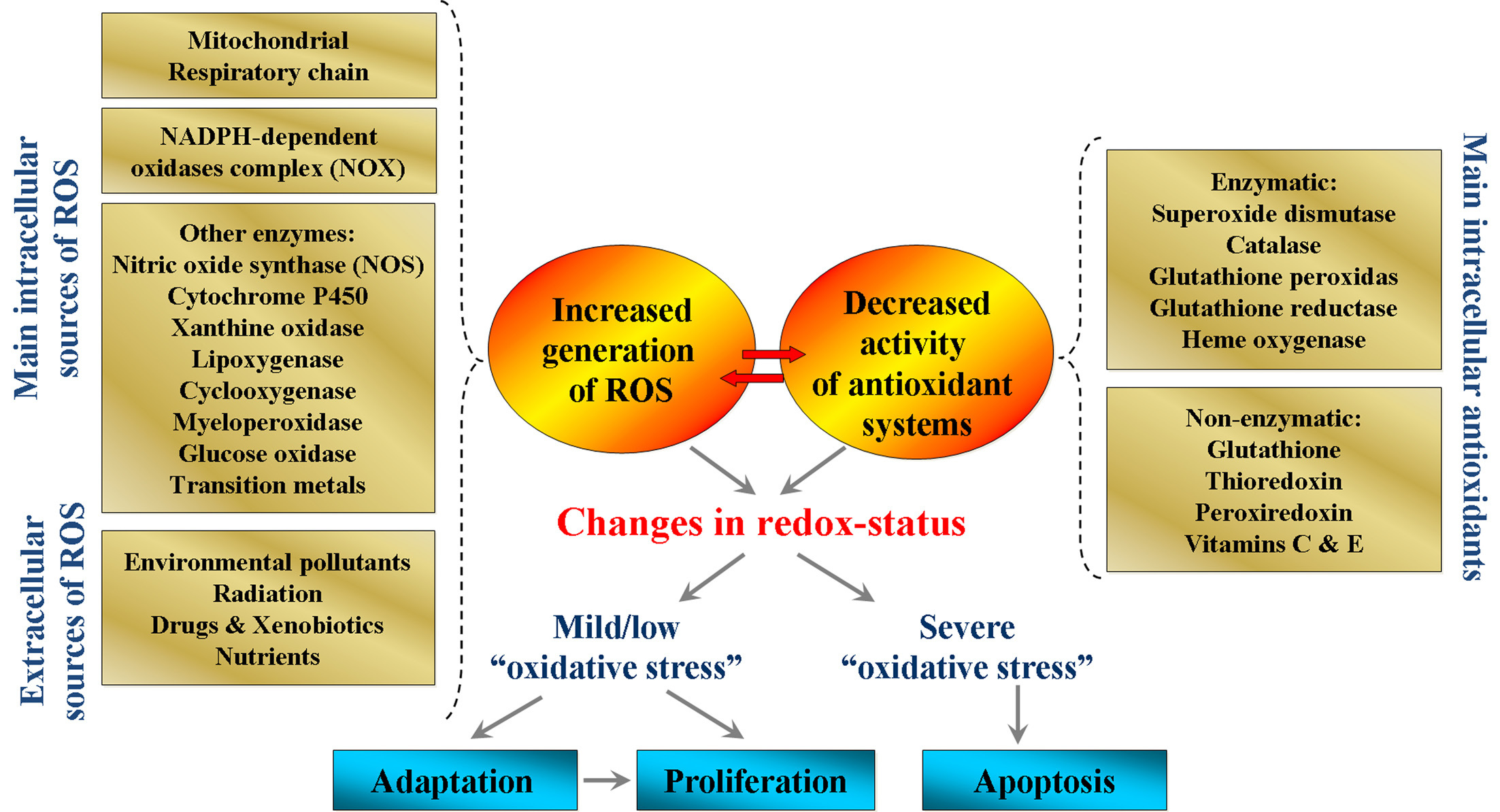
Production of ROS within certain limits is essential for the maintenance of cellular redox-homeostasis. Low/moderate levels of ROS are involved in normal biochemical pathways: 1) cellular response against infections; 2) intercellular recognition and signal transduction; and 3) induction of mitogenic response (16, 21-23). Increased intracellular levels of ROS could stimulate signal pathways, and are responsible for cell proliferation (16, 21, 22, 24-28), apoptosis (16, 22, 24-36) or activation of adaptive mechanisms through stimulation of antioxidant systems (4, 16-18, 21, 22, 24, 37-39). There are convincing evidences that low/moderate levels of ROS could activate kinases and/or inhibit phosphatases, which affect the activities of many enzymes (38, 39). The most common mechanism of these regulatory processes is the interaction of ROS with cysteine residues and formation of disulfide bonds, with subsequent activation of signal-transduction pathways (24, 35, 36, 38-40).
In contrast, overproduction of ROS above permissible levels could result in damage of the cellular macromolecules and supramolecular complexes (e.g., biomembranes) or activation of specific signaling pathways, leading to uncontrolled cell proliferation. These processes are associated with pathogenesis of different diseases, especially cancers (1, 2, 5, 8, 11, 17, 18, 21, 22, 24, 25, 27-29, 41-43). There is a hypothesis that ROS have a key role in carcinogenesis by inducing and maintaining oncogenic phenotypes in cancer cells (18, 22, 24, 36, 38, 40-44). ROS participate in the multistage carcinogenesis from initiation to malignant conversion by causing oxidative DNA damages and mutations in proto-oncogenes and tumor suppressor genes, as well as subsequent activation of signal transduction pathways (24, 38). Superoxide is considered as a main candidate among all types of ROS, which is responsible for genetic instability and malignant transformation.
Many studies suggest that cancer cells usually have an increased level of ROS, as well as over-expression of antioxidant enzymes in response to the permanent oxidative stress, in comparison with normal cells (18, 22, 24, 36, 38, 40-44). For example, Szatrowski et al. have shown that various human cancer cell lines, isolated from different tissue types, produce large amounts of hydrogen peroxide (45). Weinberg et al. have reported that defects in the manganese-dependent superoxide dismutase (Mn-SOD) induce overproduction of superoxide and cause permanent expression of cyclin D1 gene and disregulation of mitogen-activated protein kinase (MAPK) signaling pathway (10). The authors suggest that enhanced generation of ROS by cancer cells is responsible for the enhancement of their malignant (neoplastic) behavior - increasing of genomic instability and penetration into the host-tissues.
Recently, it has been found that various ROS-modulated molecular targets (signal molecules) activate different signaling pathways in the cells and could be considered as potential candidates for triggering carcinogenesis, metastasis and resistance to anticancer therapy. ROS contribute to the activation (up-regulation) of mammalian target of rapamycin (mTOR), a member of phosphoinositide 3-kinase (PI3K)-signaling pathway, which is associated with induction of malignant transformation (25, 27, 28, 46). mTOR participates in energy sensing and is a key regulator of protein synthesis, a stimulator of cell growth and proliferation. Ras proteins, which are membrane-bound G proteins with the main function of regulating cell growth and opposing apoptotic effects, are also activated by ROS and ROS-generating factors (e.g., ultraviolet irradiation, transition metals, mitogenic stimuli). It was found that the Ras gene is mutated in 30% of cancers of the lung, skin, liver, bladder and colon (47, 48). ROS cause directly mutations in tumor suppressor protein p53 (49). Mutation of p53 or loss of its function is observed in over 50% of human cancers, especially in cancers at advanced stages (50). p53 has a crucial role in sensing and removing oxidative damages of nuclear and mitochondrial DNA, preventing oxidative gene mutations and genetic instability. p53 is activated by ultraviolet irradiation, hypoxia, gamma-irradiation, and other ROS-generating factors. Several cysteine residues in central domain of p53 protein are critical for its binding to the specific DNA sequence. Since the reduction of disulphide bonds or oxidation of sulfhydryl groups often occurs at a post-translational level, p53 is considered as one of the oxidative stress response transcription factors (16). ROS induce release of calcium from intracellular stores, resulting in activation of kinases, such as protein kinases C (PK-C), which is a member of serine/threonine kinases (16). PK-C contains several cysteine-rich regions both in the zinc finger of the regulatory domain and in the catalytic site, which can be modified by various oxidants (51). One of the possible mechanisms of PK-C activation is tyrosine phosphorylation and conversion to the Ca2+/phospholipid-independent form. It seems that oxidant-induced PK-C activation plays a crucial role in cell proliferation (16, 52). It has been reported that ROS promote expression of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and activator protein 1 (AP-1) - transcription factors, that also control cytokine production and cell survival (53-55). The mechanisms of ROS-mediated expression of the transcription factors are not clear yet, but studies indicate that ROS, serve as secondary messengers, are involved in the activation of NF-κB via tumor necrosis factor (TNF) and interleukin-1 (IL-1) (56). The activation of NF-κB can be blocked by antioxidants, such as L-cysteine, N-acetyl-cysteine (NAC), other thiols, polyphenols and Vitamin E (16). Some studies demonstrate that ROS stimulate cancer cells to secrete matrix metalloproteinases (MMPs), which leads to vascular growth within the tumor microenvironment (angiogenesis) and increases the risk of blood-borne metastases (57, 58). This process is associated with ROS-mediated activation of NF-κB and AP-1 and their binding to cysteine-rich sites in the promoter regions of the MMP-2, MMP-9 and MMP-12 genes (53, 59).
All molecular mechanisms described above, as well as many other mechanisms could trigger simultaneously two opposite events - uncontrolled cell proliferation or permanent cell cycle arrest. This is the most impressive fact in these signaling pathways - any signal, for example, intracellular overproduction of ROS can induce two opposite effects: 1) activation of proliferative signaling pathways and cell survival; or 2) activation of the apoptotic signaling pathways and cell death. This phenomenon is usually explained by the different amounts of ROS. It is widely accepted that the low levels of ROS activate cell proliferation, survival and viability, which are accompanied by genetic instability - a basis of carcinogenesis. The high levels of ROS are associated with apoptosis.
There are also other opinions about the main switch between proliferation and apoptosis, but the aim of this review is to focus on one matter with practical importance - how to induce highly specific apoptosis only in cancer cells, without overproduction of ROS and harmful effects on normal cells and tissues?
ROS and involvement of redox-status in apoptosis
A large number of studies demonstrate the relationship between the increased intracellular levels of ROS and induction of apoptosis in the cells (60-65). As it was described above, the mechanisms of induction of apoptosis by most of the conventional anticancer drugs have been associated with enhanced levels of ROS and/or decreased activity of antioxidant enzymes, which is accompanied by changes in cellular redox-homeostasis (20, 29, 66-74). This observation suggests that increasing ROS and changing cellular redox-homeostasis could be part of a signal pathway, which induces apoptosis (65).
Apoptosis is a form of programmed cell death, which physiologically plays a role in embryogenesis, metamorphosis, differentiation, proliferation, as a defense mechanism to remove infected, mutated or damaged cells (75, 76). Apoptosis is physiologically more advantageous than necrosis, because the apoptotic cells are removed by phagocytosis and subsequent intracellular degradation, and thus preventing induction of inflammatory response and damage of surrounding tissue. Under normal conditions, the balance between apoptosis and cell survival is important in the development of multicellular organisms and in the regulation and maintenance of cell populations in tissues. In fact, dysfunction of the apoptotic program is implicated in a variety of pathological conditions. Thus, defects in apoptosis can result in cancer, autoimmune diseases and spread of viral infections, while neurodegenerative disorders and ischemic diseases are caused or enhanced by excessive apoptosis (76, 77).
The mechanisms of apoptosis are not fully understood, but numerous studies indicate the caspase cascade as a main key regulator of programmed cell death (78). Caspases are cysteine-containing, aspartic acid-specific proteases, which exist as zymogens in the cytoplasm, mitochondrial intermembrane space and nuclear matrix of almost all cells. The mammalian caspase family contains at least 14 members that are divided in two groups: initiator caspases and executioner caspases (79). The activation of caspases is due to apoptotic signals (such as ROS) and is associated with proteolytic cleavage of their N-terminal pro-domains, resulting in generation of small p10 and large p20 active subunits and formation of active p10/p20 tetramers (79). There are three alternative pathways for caspase activation. The two commonly accepted mechanisms for caspase activation and induction of apoptosis are: 1) intrinsic mitochondrial-mediated pathway; and 2) extrinsic Fas-mediated pathway (Figure 2). The third less well-known pathway for induction of apoptosis is the intrinsic endoplasmic reticulum-mediated pathway (77).
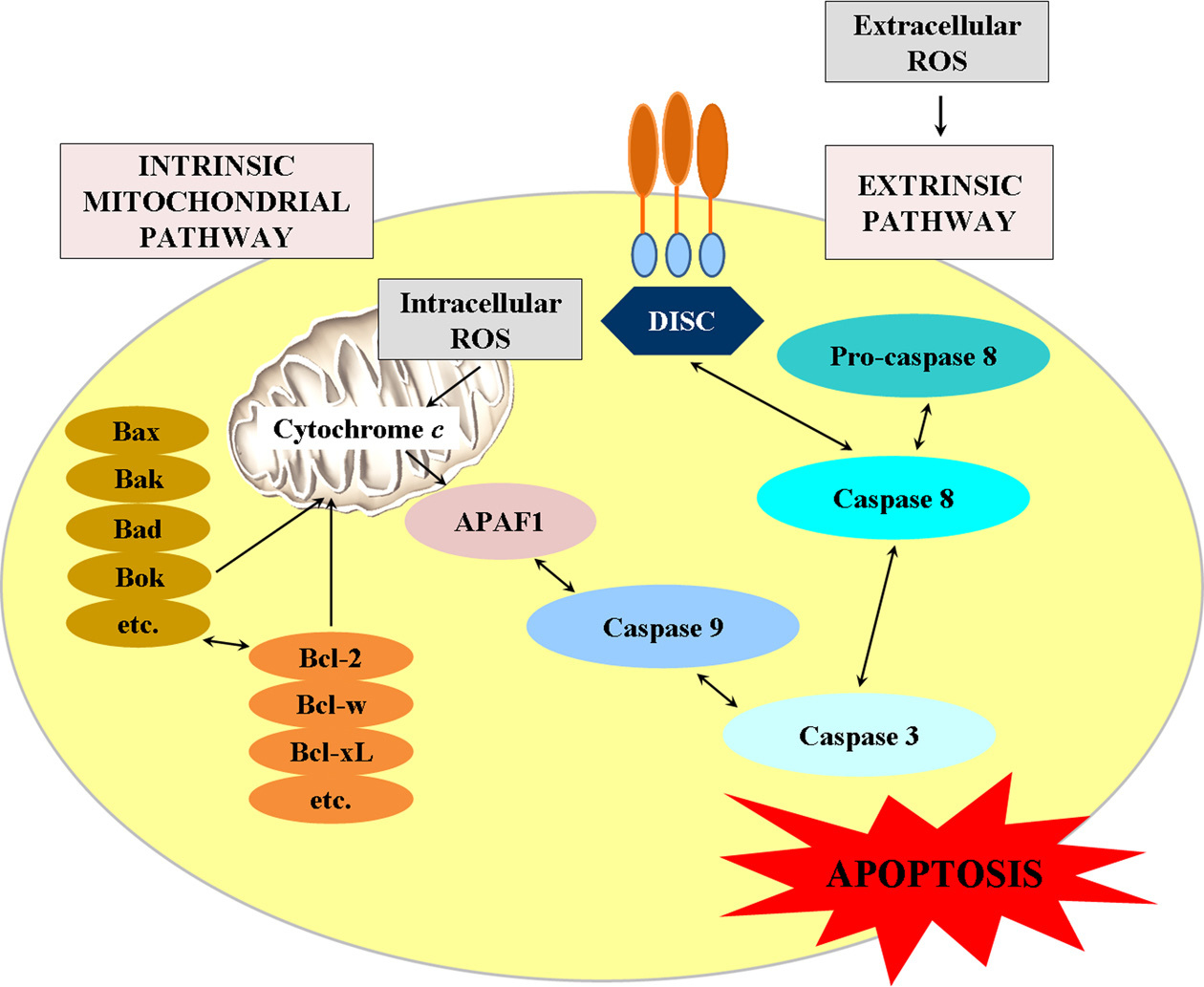
Extrinsic pathway for induction of apoptosis
The extrinsic pathway of apoptosis is mediated by death receptors. Ligand-receptor interaction initiates protein-protein interactions on cell membrane, which activates an initiator of caspases. The best known death receptors are Fas, TNF receptor type 1 (TNFR1), and receptors for TNF-related apoptosis-inducing ligand (TRAIL) type 1 and type 2 (80). The death receptors are composed of three functional extracellular domains - ligand-binding, transmembrane, and intracellular (Figure 2). The ligands, which activate death receptors, belong to the TNF superfamily of cytokines (81). Apoptotic signaling is initiated when the ligand binds to the death-receptor to form whole ligand-receptor-adaptor protein complex as the death-inducing signaling complex (DISC). Then, DISC initiates the assembly and activation of pro-caspase-8. The activated form of the enzyme (caspase-8) initiates apoptosis by cleaving other downstream caspases or executioner caspases (79).
Recent studies suggest that ROS could be a direct activator of the death receptor and apoptotic induction through formation of lipid-raft-derived signaling platforms, but the mechanism remains to be defined.
Intrinsic mitochondrial pathway for induction of apoptosis
Intrinsic pathway is initiated within the cells. Internal stimuli, such as intracellular ROS, mitochondrial DNA damage, hypoxia and extremely high concentration of cytosolic calcium ions, are triggers of the mitochondrial pathway (82). Regardless of the stimuli, this pathway is a result of increased permeability of mitochondrial membrane and release of pro-apoptotic molecules (as cytochrome c) from the intermembrane space of mitochondria into the cytosol (83, 84).
Cytochrome c is a water-soluble heme-containing protein, binding to the outer leaflet of the mitochondrial inner membrane through interactions with the anionic phospholipid cardiolipin. Normally, cytochrome c participates in the transfer of electrons between complex III and complex IV of the mitochondrial electron-transport chain (68, 85). During mitochondrial dysfunction, which is characterized by overproduction of ROS, the tightly bound cytochrome c exhibits increased peroxidase activity, oxidizes cardiolipin and facilitates its detachment (85-87). The oxidized cardiolipin is distributed to the outer part of the mitochondrial membrane and functions as a dock-platform for tBid (a membrane-targeted death ligand), which increases permeability of mitochondrial membrane and facilitates the movement of cytochrome c into the cytosol (88). Therefore, the level of cardiolipin oxidation could be an important determinant of apoptotic susceptibility of the cells, but this possibility has to be proved. It is interesting that the redox-state of cytochrome c has been implicated in mitochondrial apoptotic signaling - cytochrome c is capable to activate caspases only when it is oxidized (89, 90). In the cytosol, the release of cytochrome c can activate caspase-9, which in turn cleaves and activates executioner caspase-3. Then, some specific substances for caspase-3 [such as poly(ADP-ribose) polymerase (PARP)] could lead to apoptosis (91, 92).
The intrinsic mitochondrial pathway is regulated by proteins belonging to the B-cell lymphoma 2 (Bcl-2) family, which participate in the construction of the outer mitochondrial membrane and determine its permeability (93). There are two main groups of Bcl-2 proteins: 1) pro-apoptotic proteins (e.g., Bax, Bak, Bad, Bid, Bik, Bim) and 2) anti-apoptotic (e.g., Bcl-2, Bcl-w, Bfl-1, Bcl-xl). The anti-apoptotic proteins regulate apoptosis by blocking the mitochondrial release of cytochrome c, but the pro-apoptotic proteins act by promoting such release. It is hypothesized that the balance between pro- and anti-apoptotic proteins determines whether apoptosis would be initiated or not (93).
Many studies suggest that ROS can directly increase mitochondrial permeability and provoke depolarization of mitochondrial membrane with complete loss of mitochondrial potential (63-65, 83, 85). For example, the components of the mitochondrial permeability transition pores [e.g., voltage-dependent ion channels (DAC), adenine nucleotide translocase (ANT), cyclophilin D (cypD)] are targets of ROS and their oxidative modifications affect significantly the mitochondrial fluxes (62, 94, 95). The increased hyperpolarization of mitochondrial membrane after treatment with hydrogen peroxide initiates a collapse of the mitochondrial membrane potential and translocation of mitochondrial pro-apoptotic proteins (Bax and Bad) and cytochrome c release (62, 65, 67, 82, 84, 85, 88, 94-96).
The opening of permeability transition pores (PTPs) is considered as main event, which leads to mitochondrial depolarization and release of apoptotic factors (80). PTPs are considered as a multi-protein complex, although their exact composition is still matter of debate. Under physiological conditions, the pores allow proper passage of small molecules “in” and “out” of the mitochondria. Under apoptotic stimuli, the opening of pores may lead to the loss of cytochrome c, malfunction of the electron-transport chain and depletion of adenosine triphosphate (ATP). The outflow of apoptotic factors can trigger cell death processes. Depending on the strength and duration of the apoptotic signal, the mitochondria may either exhibit transient (reversible) alteration in transmembrane potential, or reach a “point of no return” with a massive opening of the PTPs, collapse of membrane integrity and release of apoptotic factors (80).
Induction of oxidative stress by conventional chemotherapeutics - positive or negative approach in cancer therapy
There are many conclusive evidences which have shown that conventional anticancer drugs such as anthracyclines, widely used to treat various malignant tumors, are generators of ROS, inductors of oxidative stress and initiators of apoptosis in the cells (Figure 3) (20, 66-74). For example, the anticancer activity of one of the most effective anthracycline antibiotics - doxorubicin, is due to the induction of DNA-damage, mainly through inhibition of DNA topoisomerase II enzyme after induction of double-strand DNA breaks (29). It has been found that doxorubicin is localized into the mitochondria and is involved in biochemical reactions with production of ROS and other products of free-radical oxidation, which could induce mitochondrial swelling, ultrastructural changes and mitochondrial dysfunction. Doxorubicin-induced oxidative stress causes depolarization of the mitochondrial membrane and induction of apoptosis (71, 97). Mizutani et al. have reported that doxorubicin triggers directly a production of hydrogen peroxide and induces apoptosis in human promyelocytic leukemia cell line HL-60 (66). The authors have concluded that doxorubicin-induced overproduction of hydrogen peroxide and oxidative DNA damage are the most crucial apoptotic triggers, although doxorubicin-induced apoptosis may also involve topoisomerase II inhibition (66). Tsang et al. have reported that doxorubicin increases the intracellular hydrogen peroxide, followed by mitochondrial membrane depolarization, cytochrome c release and caspase-3 activation prior to DNA laddering in p53-null human osteosarcoma Saos-2 cells (67). These processes are accompanied by up-regulation of pro-apoptotic protein Bax and down-regulation of anti-apoptotic protein Bcl-2. The authors have also established that the catalase suppresses doxorubicin-induced apoptosis by abolishing of Bax up-regulation without affecting Bcl-2 down-regulation (67). The results from this study suggest that ROS and particularly hydrogen peroxide may act as a signal molecule for doxorubicin-induced cell death even in the absence of p53 tumor suppressor protein.
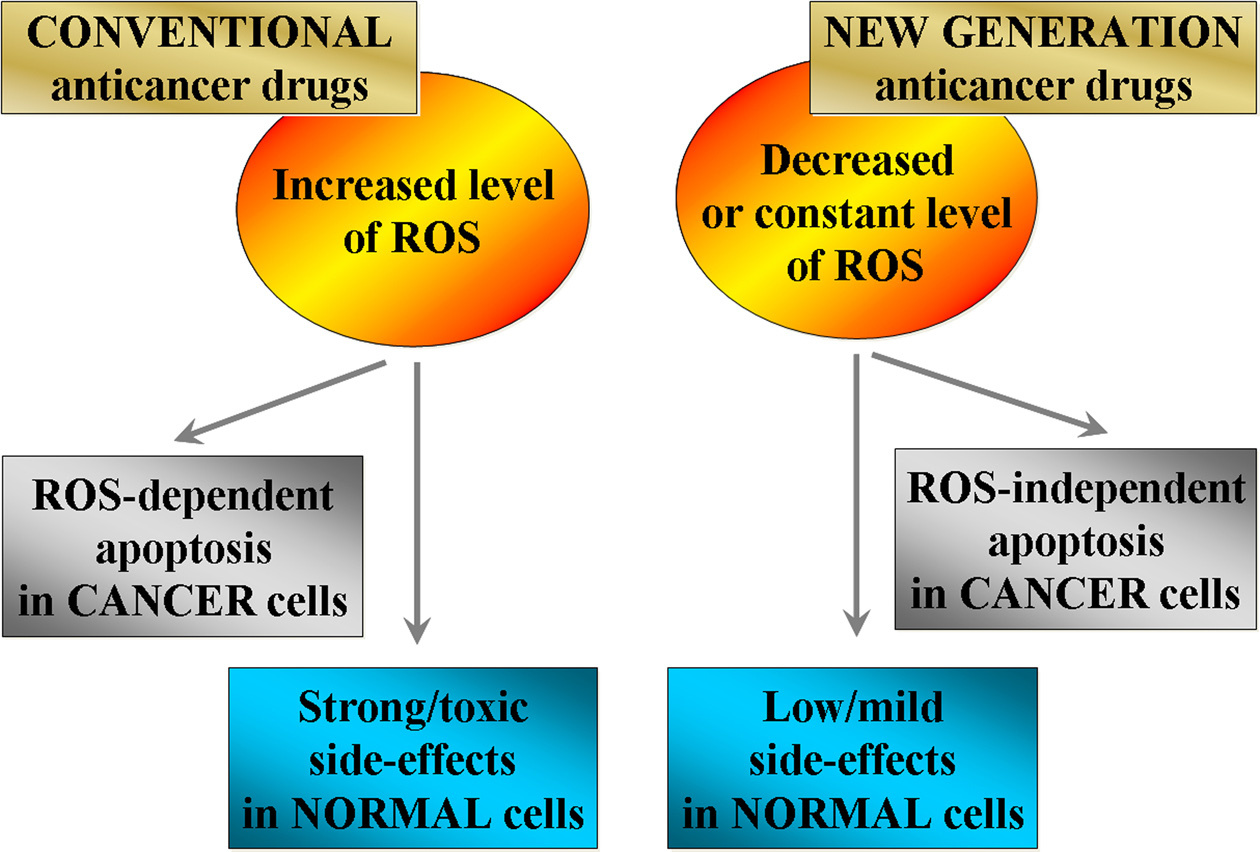
Bleomycin, also a widely used anticancer drug (conventional) for treatment of lymphomas, squamous cell carcinomas and testicular tumors, causes single- and double-strand breaks in DNA in vivo and in vitro, which results in cell damage (72, 98-100). The mechanism of the antineoplastic effect of bleomycin involves the formation of bleomycin-iron complex, which can reduce molecular oxygen to superoxide and hydroxyl radicals that can attack DNA and trigger its cleavage (72). Wallach-Dayan et al. have reported that bleomycin increases ROS in mouse lung epithelial (MLE) cells, which is accompanied by mitochondrial leakage, caspase-8 and caspase-9 activation, up-regulation of apoptotic (tumor necrotic) factor Fas, and induction of apoptosis (99). Glutathione inhibits these effects, which suggests their dependence on ROS and particularly on hydrogen peroxide. In the study conducted by Cort et al., human testicular cancer cells are incubated with bleomycin or hydrogen peroxide for 72 h (100). The authors have observed that both substances induce apoptosis by enhancement of the caspase activity, release of cytochrome c into the cytosol, increase of Bax level and decrease of Bcl-2 level.
Cisplatin is another widely used anticancer drug, generally recognized as DNA-damaging agent (101). It interacts predominantly with nucleophilic N7-sites of purine bases of DNA, leading to formation of DNA-protein and DNA-DNA inter-strand and intra-strand cross-links (102). It has been shown that cisplatin induces production of ROS, as well as interacts with mitochondrial DNA (101, 103). Harhaji-Trajkovic et al. have reported that cisplatin increases ROS level and causes caspase activation and DNA fragmentation in U251 glioma, C6 glioma and L929 fibrosarcoma cell lines (102). Bragodo et al. have established on HCT116 colon carcinoma-derived cells that the apoptotic activity of cisplatin requires the onset of a p53-mediated p38α MAPK-pathway through generation of ROS (104). All these studies suggest ROS-mediated induction of apoptosis by cisplatin, but do not specify which types of ROS are responsible for this process.
Some of the most effective chemotherapeutics, which are used for treatment of variety of malignancies, have shown toxic side effects on non-cancer tissues. For example, the side effects of doxorubicin are attributed to chronic toxic effects in the heart as cardiomyopathy and congestive heart failure (29, 69-71, 97), those of bleomycin are associated with induction of pulmonary fibrosis (72, 99), and those of cisplatin are kidney damages, gastrointestinal dysfunction and neuropathy (73, 103). The side effects of conventional anticancer drugs could be also due to intracellular chelation of transition metals (e.g., iron and/or cupper ions) and triggering of Fenton-type reactions with subsequent production of the highly reactive hydroxyl radicals (19, 105, 106). All studies mentioned above, as well as many other studies consider that the efficiency of the conventional anticancer drugs is due to induction of oxidative stress in the cancer cells, but they also suggest that this is a reason for their toxic side effects on non-cancer cell and tissues due to induction of redox-imbalance in whole organism (30, 66, 67, 69-74, 97-99, 103, 106, 107).
Currently, it is not clear whether the mechanisms for induction of ROS by conventional anticancer drugs are the same in cancer and non-cancer cells or they are different. It is generally accepted that the accumulation of genetic alterations and mutations in cancer cells are the main triggers of ROS. In non-cancer cells, the mechanisms might be different, e.g., by induction of endoplasmic reticulum stress and subsequent production of ROS (108-111). Thus, the efforts of scientists are directed to achieve therapeutic selectivity and prevent side effects and drug resistance by altering the unique biochemical pathways that distinguish the cancer cells from normal ones and to design new therapeutic strategies based on their different redox-homeostasis (19, 110). It raises several questions: 1) Is it possible to develop therapeutic approach, accompanied by induction of apoptosis in cancer cells without changes in the redox-homeostasis of the normal cells? 2) Is it possible to induce apoptosis in cancer cells by ROS-independent mechanisms? 3) Which is better to increase or decrease ROS in cancer cells?
Is it obligatory to raise ROS level to induce apoptosis in cancer cells?
ROS as a consequence (side effect) of apoptosis
It is still disputable whether the ROS are a reason for induction of apoptosis or are they a side effect induced by the mechanism of cell death. The initial idea for the role of ROS in apoptosis was based on the observation that Bcl-2 has an apparent antioxidant function in mammalian cells (111). Experimental data have shown that over-expression of Bcl-2 protects cells from lipid peroxidation and thiol oxidation, induced by menadione and hydrogen peroxide (111-113). Bcl-2 also protects cells from apoptosis at limited production of ROS. For example, Cai et al. have used staurosporine-treated HL-60 cells to study the mechanism of generation of ROS during apoptosis (111). The authors try to clarify whether the oxidation signals in the cells precede or follow the release of cytochrome c into the cytosol and initiation of apoptosis. They measure the dynamic of the redox-state of intracellular glutathione as a function of the incubation time with staurosporine (an inductor of apoptosis). The results have demonstrated a release of cytochrome c from mitochondria into the cytosol after 1-hour incubation with staurosporine, while the oxidation of intracellular glutathione begins after 2-hours incubation (111). Usually antioxidants, such as N-acetyl-cysteine, can inhibit apoptosis in cells, whose mechanism for induction of cell death depends on their redox-status (114). However, the experiments of Cai et al. show that in staurosporine-induced apoptosis, the caspase activation seems to be redox-insensitive since the treated cells are not sensitive to N-acetyl-cysteine. These results are also confirmed by measurement of DNA fragmentation and phosphatidylserine translocation on the surface of cell membrane (111).
In vitro data on cell-free systems have indicated that the caspase 3 activation is independent on the redox-state of cytochrome c and the activation of caspase 3 and caspase 9 could cause ROS production (111, 115). Thus, when ROS is generated simultaneously with the direct activation of caspases, it might be only a side effect, which does not relate to the key signaling events leading to the caspase-dependent cell death (111).
ROS-independent apoptosis
Recently, several studies have reported induction apoptosis by ROS-independent mechanism (116-120). Jacobson and Raff have shown that apoptosis has occurred when the cells are cultivated in hypoxic conditions without presence of ROS-generating substances, such as menadione or hydrogen peroxide (116). In similar experiments, Shimizu et al. have failed to detect ROS during hypoxia-induced apoptosis (117). These evidences suggest that ROS are not obligator effectors in the apoptosis. Ko et al. have also described ROS-independent cell death in HL-60 cells, induced by myricetin, a flavonoid with antioxidant properties (118). In this study, the myricetin-inducing apoptosis is characterized by decrease of mitochondrial functions and Bcl-2/Bax ratio, translocation of cytochrome c from the mitochondria into the cytosol and activation of caspases 3 and 9 without significant induction of intracellular ROS. ROS-independent mechanism of apoptosis in myricetin-treated HL-60 cells has confirmed by using antioxidants such as N-acetyl-cysteine, catalase, SOD, and tiron. All antioxidants and antioxidant enzymes do not protect the myricitin-treated cells (118). Furthermore, the authors have observed that myricetin significantly decreases hydrogen peroxide-induced intracellular production of hydroperoxides (118). Hou et al. have demonstrated ROS-independent cell death after treatment of HL-60 cells with gossypol, a polyphenolic compound existing in the seeds of cotton (119). The level of ROS in gossypol-treated cells was similar to that in non-treated cells. Antioxidants, such as N-acetyl-cysteine and catalase, did not inhibit gossypol-induced caspase-3 activation, PARP degradation, and DNA fragmentation (119). The gossypol-induced apoptosis was accompanied by cleavage of Bid protein, loss of mitochondrial potential, cytochrome c release into the cytosol, and activation of caspases 3, 8, and 9 (119). Similar data have been reported by Lin et al. (120). The authors have observed that rhein, an anthraquinone compound used in Chinese medicine, induces apoptosis in HL-60 cells by ROS-independent mechanism(s), because the antioxidants such as N-acetyl-cysteine, tiron and catalase did not suppress rhein-induced apoptosis (120).
Combination between redox-modulators and anticancer drugs as a possible trigger of ROS-independent apoptosis in cancer cells
In the last two decades, many data have shown that natural or synthetic compounds, which modulate cellular redox-status, exhibit also anticancer effects (80, 121-126). Some of these redox-modulators could induce production of ROS, but in most cases their anticancer activity is connected to the influence of signaling pathways for regulation of physiological processes (e.g., pathways responsible for cell survival, proliferation, or apoptosis), rather than strong oxidative stress and damage of biomacromolecules. For example, the anticancer effect of 2-deoxy-D-glucose (2-DDG), an inhibitor of glycolysis, is connected with intracellular ATP depletion (127), disruption of thiol metabolism (128-130), limited synthesis of NADPH (50% in comparison with non-treated cells) (131) and eventually induction of oxidative stress mainly in the cancer cells (112, 129, 131).
Aykin-Burns et al. have studied the sensitivity of cancer cells and normal cells to glucose-deprivation-induced cytotoxicity (132). The authors have measured the levels of superoxide and hydroperoxides inducing by treatment of both types of cells with 2-DDG. The results have shown that cancer cells are characterized by significantly increased levels of superoxide (2-20-fold) and hydroperoxides (1.8-10-fold), in comparison with the normal cells (132).
In our recent study, we also found that 2-DDG sensitizes leukemia cells (Jurkat, derived from acute lymphoblastic leukemia) to new generation anticancer drugs, such as barasertib, bortezomib, everolimus, lonafarnib and palbociclib (132). In the case of everolimus and barasertib, the combination with 2-DDG leads to a very strong synergistic cytotoxicity, accompanied by a strong induction of apoptosis, without increasing ROS level in the treated cells. In contrast, 2-DDG decreases doxorubicin-mediated generation of ROS and induction of apoptosis, which results in diminishing of the cytotoxicity of doxorubicin towards those cells. This study shows that combining new-generation anticancer drugs with a redox-modulator (such as 2-DDG) markedly enhances the anticancer effect at a very low concentration of the drug.
Many in vitro and in vivo studies suggest that other redox-modulator, docosahexaenoic acid (DHA: an essential fatty acid derived from fish oil), also has anticancer activity and can improve the efficiency of conventional cancer therapy: DHA suppresses tumor cell proliferation, reduces tumor growth in experimental animals, inhibits drug resistance in various cancer cell lines, and exerts cytotoxic effects on cancer cells, but it is significantly less cytotoxic for normal cells (133-142).
Some authors have observed that the combination of DHA with anticancer drugs induces apoptosis in cancer cells, which is accompanied by an enhancement of the level of ROS (143-145), but others did not detect any changes in this parameter (146, 147). For example, Lin et al. have found that DHA inhibits doxorubicin-induced generation of ROS and elevates the activity of catalase and SOD in NRK-52E cells, a rat renal proximal tubular cell line (147). DHA sensitizes cancer cell lines A-172 and U-87 MG (derived from glioblastoma) and A-427 (derived from bronchial carcinoma) to doxorubicin (146). In this case, DHA increases the level of ROS in doxorubicin-treated A-427 cells, but does not influence the level of ROS in A-172 and U-87 cells.
Currently, we described a synergistic cytotoxicity between DHA and anticancer drugs everolimus and barasertib, which is ROS-dependent, but specific for cancer cells (particularly for Jurkat) (148). These combinations were harmless to normal lymphocytes and did not induce abnormal production of ROS in those cells. Our data suggest that DHA could be used as a supplementary component in the anticancer chemotherapy, which allows decreasing the therapeutic doses of everolimus and barasertib and minimizing their side effects. This assumption can also be made for other chemotherapeutics that show sinergistic cytotoxic effects in combination with DHA (e.g., palbociclib and lonafarnib).
Conclusions
The data described above suggest that in some cases using a redox-modulator, it is possible to influence the cellular redox-status in such a way in order to reduce the production of ROS, and to induce apoptosis in cancer cells by ROS-independent mechanisms. This could be a possible approach to minimize side effects of conventional cancer therapy on normal cells and tissues.
Acknowledgements
None.
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev 2000;80:315–60. [PubMed]
- Macmillan-Crow LA, Cruthirds DL. Invited review: manganese superoxide dismutase in disease. Free Radic Res 2001;34:325–36. [PubMed] DOI:10.1080/10715760100300281
- Narvaez CJ, Welsh J. Role of mitochondria and caspases in vitamin D-mediated apoptosis of MCF-7 breast cancer cells. J Biol Chem 2001;276:9101–7. [PubMed] DOI:10.1074/jbc.M006876200
- Van Remmen H, Ikeno Y, Hamilton M, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics 2003;16:29–37. [PubMed] DOI:10.1152/physiolgenomics.00122.2003
- Cave AC, Brewer AC, Narayanapanicker A, et al. NADPH oxidases in cerdiovascular health and disease. Antioxid Redox Signal 2006;8:691–728. [PubMed] DOI:10.1089/ars.2006.8.691
- Lambeth JD, Krause KH, Clark RA. NOX enzymes as novel targets for drug development. Semin Immuno-pathol 2008;30:339–63. DOI:10.1007/s00281-008-0123-6
- Nauseef WM. Biological roles for the NOX family NADPH oxidases. J Biol Chem 2008;283:16961–5. [PubMed] DOI:10.1074/jbc.R700045200
- Pulkes T, Hanna MG. Human mitochondrial DNA diseases. Adv Drug Deliv Rev 2001;49:27–43. [PubMed] DOI:10.1016/S0169-409X(01)00124-7
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 2003;552:335–44. [PubMed] DOI:10.1113/jphysiol.2003.049478
- Weinberg F, Chandel NS. Reactive oxygen species-development signaling regulates cancer. Cell Mol Life Sci 2009;66:3663–73. [PubMed] DOI:10.1007/s00018-009-0099-y
- Lin TK, Liou CW, Chen SD, et al. Mitochondrial dysfunction and biogenesis in the pathogenesis of Parkinson’s disease. Chang Gung Med J 2009;32:589–99. [PubMed]
- Bedard K, Krause KH. The NOX family of ROS-genersting NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245–313. [PubMed] DOI:10.1152/physrev.00044.2005
- Van Bogaert IN, Groeneboer S, Saerens K, et al. The role of cytochrome P450 monooxygenases in microbial fatty acid metabolism. FEBS J 2011;278:206–21. [PubMed] DOI:10.1111/j.1742-4658.2010.07949.x
- Urlacher VB, Girhard M. Cytochrome P450 mono-oxygenases: an update on perspectives for synthetic application. Trends Biotechnol 2012;30:26–36. [PubMed] DOI:10.1016/j.tibtech.2011.06.012
- Wassmann S, Wassmann K, Nickenig G. Modulation of oxidant and antioxidant enzyme expression and function in vascular cells. Hypertension 2004;44:381–6. [PubMed] DOI:10.1161/01.HYP.0000142232.29764.a7
- Valko M, Leibfritz D, Moncol J, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 2007;39:44–84. [PubMed] DOI:10.1016/j.biocel.2006.07.001
- Finkel T, Holbook NJ. Oxidants, oxidative stress and the biology of ageing. Nature 2000;408:239–47. [PubMed] DOI:10.1038/35041687
- Khan MA, Tania M, Zhang DZ, et al. Antioxidant enzymes and cancer. Chin J Cancer Res 2010;22:87–92. DOI:10.1007/s11670-010-0087-7
- Morier-Teissier E, Bernier JL, Lohez M, et al. Free radical production and DNA cleavage by copper chelating peptide-anthraquinones. Anticancer Drug Des 1990;5:291–305. [PubMed]
- Wang J, Yi J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol Ther 2008;7:1875–84. [PubMed] DOI:10.4161/cbt.7.12.7067
- Sarsour EH, Venkataraman S, Kalen AL, et al. Manganese superoxide dismutase activity regulates tranitions between quiescent and proliferative growth. Aging Cell 2008;7:405–17. [PubMed] DOI:10.1111/j.1474-9726.2008.00384.x
- Kim B, Song YS. Mitochondrial dynamics altered by oxidative stress in cancer. Free Radic Res 2016:1-16. [Epub ahead of print]
- Sena LA, Chandel NS. Physiological roles of mito-chondrial reactive oxygen species. Mol Cell 2012;48:158–67. [PubMed] DOI:10.1016/j.molcel.2012.09.025
- Kahlos K, Soini Y, Pääkkö P, et al. Proliferation, apoptosis, and manganese superoxide dismutase in malignant mesothelioma. Int J Cancer 2000;88:37–43. [PubMed] DOI:10.1002/(ISSN)1097-0215
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell 2006;124:471–84. [PubMed] DOI:10.1016/j.cell.2006.01.016
- Ambrose M, Gatti RA. Pathogenesis of ataxia-telangiectasia: the next generation of ATM functions. Blood 2013;121:4036–45. [PubMed] DOI:10.1182/blood-2012-09-456897
- Alexander A, Cai SL, Kim J, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci USA 2010;107:4153–8. [PubMed] DOI:10.1073/pnas.0913860107
- Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet 2005;37:19–24. [PubMed] DOI:10.1038/ng1494
- Ozben T. Oxidative stress and apoptosis: impact on cancer therapy. J Pharm Sci 2007;96:2181–96. [PubMed] DOI:10.1002/jps.20874
- Lee YJ, Shacter E. Oxidative stress inhibits aoptosis in human lymphoma cells. J Biol Chem 1999;274:19792–8. [PubMed] DOI:10.1074/jbc.274.28.19792
- Haddad JJ. Redox and oxidant-mediated regulation of apoptosis signaling pathways: immuno-pharmaco-redox conception of oxidative siege versus cell death commitment. Int Immunopharmacol 2004;4:475–93. [PubMed] DOI:10.1016/j.intimp.2004.02.002
- Kannan K, Jain SK. Oxidative stress and apoptosis. Pathophysiology 2000;7:153–63. [PubMed] DOI:10.1016/S0928-4680(00)00053-5
- Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000;5:415–8. [PubMed] DOI:10.1023/A:1009616228304
- Salazar-Ramiro A, Ramirez-Ortega D, Pérez de la Cruz V, et al. Role of redox status in development of glioblastoma. Front Immunol 2016;7:156. [PubMed]
- Trachootham D, Zhou Y, Zhang H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006;10:241–52. [PubMed] DOI:10.1016/j.ccr.2006.08.009
- Zhou Y, Hileman EO, Plunkett W, et al. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood 2003;101:4098–104. [PubMed] DOI:10.1182/blood-2002-08-2512
- Young TW, Mei FC, Yang G, et al. Activation of antioxidant pathways in ras-mediated oncogenic transformation of human surface ovarian epithelial cells revealed by functional proteomics and mass spectrometry. Cancer Res 2004;64:4577–84. [PubMed] DOI:10.1158/0008-5472.CAN-04-0222
- Trachootham D, Lu W, Ogasawara MA, et al. Redox regulation and cell survival. Antioxid Redox Signal 2008;10:1343–74. [PubMed] DOI:10.1089/ars.2007.1957
- Penney RB, Roy D. Thioredoxin-mediated redox regula-tion of resistance to endocrine therapy in breast cancer. Biochim Biophys Acta 2013;1836:60–79. [PubMed]
- Coleman WB, Tsongalis GJ. Molecular mechanisms of human carcinogenesis. EXS 2006: 321–49. [PubMed]
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011;11:85–95. [PubMed] DOI:10.1038/nrc2981
- Zhelev Z, Aoki I, Gadjeva V, et al. Tissue redox activity as a sensing platform for imaging of cancer based on nitroxide redox cycle. Eur J Cancer 2013;49:1467–78. [PubMed] DOI:10.1016/j.ejca.2012.10.026
- Bakalova R, Zhelev Z, Aoki I, et al. Tissue redox activity as a hollmark of cancinogenesis: from early to terminal stages of cancer. Clin Cancer Res 2013;19:2503–17. [PubMed] DOI:10.1158/1078-0432.CCR-12-3726
- Suzuki K, Matsubara H. Recent advances in p53 research and cancer treatment. J Biomed Biotechnol 2011;2011:978312. [PubMed]
- Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res 1991;51:794–8. [PubMed]
- Zhang X, Cheng X, Yu L, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun 2016;7:12109. [PubMed] DOI:10.1038/ncomms12109
- Vachtenheim J. Occurrence of ras mutation in human lung cancer. Minireview. Neoplasma 1997;44:145–9. [PubMed]
- Wei M, Wanibuchi H, Morimura K, et al. Carcinogenicity of dimethylarsinic acid in male F344 rats and genetic alterations in induced urinary bladder tumors. Carcinogenesis 2002;23:1387–97. [PubMed] DOI:10.1093/carcin/23.8.1387
- Hofseth LJ, Hussain SP, Harris CC. p53:25 years after its discovery. Trends Pharmacol Sci 2004;25:177–81. [PubMed] DOI:10.1016/j.tips.2004.02.009
- Hollstein M, Sidransky D, Vogelstein B, et al. p53 mutation in human cancer. Science 1991;253:49–53. [PubMed] DOI:10.1126/science.1905840
- Gopalakrishna R, Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med 2000;28:1349–61. [PubMed] DOI:10.1016/S0891-5849(00)00221-5
- Dempsey EC, Newton AC, Mochly-Rosen D, et al. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol 2000;279:L429–38. [PubMed]
- Yoshida M, Korfhagen TR, Whitsett JA. Surfactant protein D regulates NF-kappa B and matrix metalloproteinase production in alveolar macrophages via oxidant-sensitive pathways. J Immunol 2001;166:7514–9. [PubMed] DOI:10.4049/jimmunol.166.12.7514
- Meister M, Tomasovic A, Banning A, et al. Mitogen-activated protein (MAP) kinase scaffolding proteins: a recount. Int J Mol Sci 2013;14:4854–84. [PubMed] DOI:10.3390/ijms14034854
- Dhar A, Young MR, Colburn NH. The role of AR-1, NF-kappaB and ROS/NOS in skin carcinogenesis: the JB6 model is predictive. Mol Cell Biochem 2002;234-235:185–93. [PubMed] DOI:10.1023/A:1015948505117
- Hughes G, Murphy MP, Ledgerwood EC. Mitochondrial reactive oxygen species regulate the temporal activation of nuclear factor-kappaB to modulate tumor necrosis factor-induced apoptosis: evidence from mitochondria-targeted antioxidants. Biochem J 2005;389:83–9. [PubMed] DOI:10.1042/BJ20050078
- Storz P. Reactive oxygen species in tumor progression. Front Biosci 2005;10:1881–96. [PubMed] DOI:10.2741/1667
- Zhang HJ, Zhao W, Venkataraman S, et al. Activation of matrix metalloproteinase-2 by overexpression of manganese superoxide dismutase in human breast cancer MCF-7 cells involves reactive oxygen species. J Biol Chem 2002;277:20919–26. [PubMed] DOI:10.1074/jbc.M109801200
- Harendza S, Pollock AS, Mertens PR, et al. Tissue-specific enhancer-promoter interactions regulate high level constitutive expression of matrix metalloproteinase 2 by glomerular mesangial cells. J Biol Chem 1995;270:18786–96. [PubMed] DOI:10.1074/jbc.270.32.18786
- Lennon SV, Martin SJ, Cotter TG. Dose-dependent induction of apoptosis in human tumour cell lines by widely diverging stimuli. Cell Prolif 1991;24:203–14. [PubMed] DOI:10.1111/j.1365-2184.1991.tb01150.x
- Fernandes RS, Cotter TG. Apoptosis or necrosis: intracellular levels of glutathione influence mode of cell death. Biochem Pharmacol 1994;48:675–81. [PubMed] DOI:10.1016/0006-2952(94)90044-2
- Hockenbery DM, Oltvai ZN, Yin XM, et al. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 1993;75:241–51. [PubMed] DOI:10.1016/0092-8674(93)80066-N
- Torres-Roca JF, Lecoeur H, Armatore C, et al. The early intracellular production of reactive oxygen intermediates mediates apoptosis in dexamethasone-treated thymocytes. Cell Death Differ 1995;2:309–19. [PubMed]
- Forrest VJ, Kang YH, McClain DE, et al. Oxidative stress-induced apoptosis prevented by Trolox. Free Radic Biol Med 1994;16:675–84. [PubMed] DOI:10.1016/0891-5849(94)90182-1
- Higuchi M, Honda T, Proske RJ, et al. Regulation of reactive oxygen species-induced apoptosis and necrosis by caspase 3-like proteases. Oncogene 1998;17:2753–60. [PubMed] DOI:10.1038/sj.onc.1202211
- Mizutani H, Tada-Oikawa S, Hiraku Y, et al. Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide. Life Sci 2005;76:1439–53. [PubMed] DOI:10.1016/j.lfs.2004.05.040
- Tsang WP, Chau SP, Kong SK, et al. Reactive oxygen species mediate doxorubicin induced p53-independent apoptosis. Life Sci 2003;73:2047–58. [PubMed] DOI:10.1016/S0024-3205(03)00566-6
- Tsai SY, Sun NK, Lu HP, et al. Involment of reactive oxygen species in multidrug resistance of vincristine-selected lymphoblastoma. Cancer Sci 2007;98:1206–14. [PubMed] DOI:10.1111/cas.2007.98.issue-8
- Salvatorelli E, Guarnieri S, Menna P, et al. Defective one- or two-electron reduction of the anticancer anthracycline epirubicin in human heart. J Biol Chem 2006;281:10990–1001. [PubMed] DOI:10.1074/jbc.M508343200
- den Hartog GJ, Haenen GR, Boven E, et al. Lecithinized copper, zinc-superoxide dismutase as a protector against doxorubicin-induced cardiotoxicity in mice. Toxicol Appl Pharmacol 2004;194:180–8. [PubMed] DOI:10.1016/j.taap.2003.09.008
- Konorev EA, Kennedy MC, Kalyanaraman B. Cell-permeable superoxide dismutase and glutathione peroxidase mimetics afford superior protection against doxorubicin-induced cardiotoxicity: the role of reactive oxygen and nitrogen intermediates. Arch Biochem Biophys 1999;368:421–8. [PubMed] DOI:10.1006/abbi.1999.1337
- Hagiwara SI, Ishii Y, Kitamura S. Aerosolized adminis- tration of N-acetylcysteine attenuates lung fibrosis induced by bleomycin in mice. Am J Respir Crit Care Med 2000;162:225–31. [PubMed] DOI:10.1164/ajrccm.162.1.9903129
- Satoh M, Kashihara N, Fujimoto S, et al. A novel free radical scavenger, edarabone, protects against cisplatin-induced acute rental damage in vitro and in vivo. J Pharmacol Exp Ther 2003;305:1183–90. [PubMed] DOI:10.1124/jpet.102.047522
- Hwang JT, Ha J, Park OJ. Combination of 5-fluorouracil and genistein induces apoptosis synergistically in chemo-resistant cancer cells through the modulation of AMPK and COX-2 signaling pathways. Biochem Biophys Res Commun 2005;332:433–40. [PubMed] DOI:10.1016/j.bbrc.2005.04.143
- Morales-González JA, Cardoso AB, Rodríguez FM, et al. Programmed cell death (apoptosis): the regulating mechanisms of cellular proliferation. Arch Neurocien 2004;9:85–93.
- León J, Acu?a-Castroviejo D, Escames G, et al. Melatonin mitigates mitochondrial malfunction. J Pineal Res 2005;38:1–9. [PubMed] DOI:10.1111/jpi.2005.38.issue-1
- Fadeel B, Orrenius S, Zhivotovsky B. Apoptosis in human disease: a new skin for the old ceremony. Biochem Biophys Res Commun 1999;266:688–717.
- Ogata S, Takeuchi M, Fujita H, et al. Apoptosis induced by nicotinamide-related compounds and quinolinic acid in HL-60 cells. Biosci Biotachnol Biochem 2000;64:327–32. DOI:10.1271/bbb.64.327
- Nicholson DW. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ 1999;6:1028–42. [PubMed] DOI:10.1038/sj.cdd.4400598
- Berg D, Lehne M, Müller N, et al. Enforced covalent trimerization increases the activity of the TNF ligand family members TRAIL and CD95L. Cell Death Differ 2007;14:2021–34. [PubMed] DOI:10.1038/sj.cdd.4402213
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell 2004;116:205–19. [PubMed] DOI:10.1016/S0092-8674(04)00046-7
- O'Brien MA, Kirby R. Apoptosis: A review of pro-apoptotic and anti-apoptotic pathways and dysregulation in disease. J Vet Emerg Crit Care 2008;18:572–85. DOI:10.1111/vec.2008.18.issue-6
- Park MT, Kim MJ, Kang YH, et al. Phytosphingosin in combination with ionizing radiation enhances apoptotic cell death in radiation-resistant cancer cells through ROS-dependent and -independent AIF release. Blood 2005;105:1724–33. [PubMed] DOI:10.1182/blood-2004-07-2938
- Susin SA, Lorenzo HK, Zamzami N, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999;397:441–6. [PubMed] DOI:10.1038/17135
- Ott M, Robertson JD, Gogvadze V, et al. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA 2002;99:1259–63. [PubMed] DOI:10.1073/pnas.241655498
- Kagan VE, Borisenko GG, Tyurina YY, et al. Oxidative lipidomics of apoptosis: redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic Biol Med 2004;37:1963–85. [PubMed] DOI:10.1016/j.freeradbiomed.2004.08.016
- Dejean LM, Martinez-Caballero S, Kinnally KW. Is MAC the knife that cuts cytochrome c from mitochondria during apoptosis. Cell Death Differ 2006;13:1387–95. [PubMed] DOI:10.1038/sj.cdd.4401949
- Gonzalvez F, Pariselli F, Dupaigne P, et al. tBid interaction with cardiolipin primarily orchestrates mitochondrial dysfunctions and subsequently activates Bax and Bak. Cell Death Differ 2005;12:614–26. [PubMed] DOI:10.1038/sj.cdd.4401571
- Brown GC, Borutaite V. Regulation of apoptosis by the redox state of cytochrome c. Biochim Biophys Acta 2008;1777:877–81. [PubMed] DOI:10.1016/j.bbabio.2008.03.024
- Suto D, Sato K, Ohba Y, et al. Suppression of the pro-apoptotic function of cytochrome c by singlet oxygen via a haem redox state-independent mechanism. Biochem J 2005;392:399–406. [PubMed] DOI:10.1042/BJ20050580
- Hou DX, Tong X, Terahara N, et al. Delphinidin3-sambubioside, a Hibiscus anthocyanin, induces apoptosis in human leukemia cells through reactive oxygen species-mediated mitochondrial pathway. Arch Biochem Biophys 2005;440:101–9. [PubMed] DOI:10.1016/j.abb.2005.06.002
- Ogata S, Takeuchi M, Fujita H, et al. Apoptosis induced by nicotinamide-related compounds and quinolinic acid in HL-60 cells. Biosci Biotachnol Biochem 2000;64:327–32. DOI:10.1271/bbb.64.327
- Ryu SW, Kim E. Apoptosis induced by human Fas-associated factor 1, hFAF1, requires its ubiquitin homologues domain, but not the Fas-binding domain. Biochem Biophys Res Commun 2001;286:1027–32. [PubMed] DOI:10.1006/bbrc.2001.5505
- Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 2007;12:835–40. [PubMed] DOI:10.1007/s10495-006-0525-7
- Madesh M, Hajnóczky G. VDAC-dependent permeabili-zation of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J Cell Biol 2001;155:1003–15. [PubMed] DOI:10.1083/jcb.200105057
- Skulachev VP. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 2006;11:473–85. [PubMed] DOI:10.1007/s10495-006-5881-9
- Danz ED, Skramsted J, Henry N, et al. Resveratrol prevents doxorubicin cardiotoxicity through mito-chondrial stabilization and Sirt1 pathway. Free Radic Biol Med 2009;46:1589–97. [PubMed] DOI:10.1016/j.freeradbiomed.2009.03.011
- Kasper M, Barth K. Bleomycin and its role in inducting apoptosis and senescence in lung cells-modulating effects of caveolin-1. Curr Cancer Drug Targets 2009;9:341–53. [PubMed] DOI:10.2174/156800909788166501
- Wallach-Dayan SB, Izbicki G, Cohen PY, et al. Bleomycin initiates apoptosis of lung epithelial cells by ROS but not by Fas/FasL pathway. Am J Physiol Lung Cell Mol Physiol 2006;290:L790–6. [PubMed] DOI:10.1152/ajplung.00300.2004
- Cort A, Timur M, Dursun E, et al. Effect of N-acetylcystein on bleomycin-induced apoptosis in malignant testicular germ cell tumors. J Physiol Biochem 2012;68:555–62. [PubMed] DOI:10.1007/s13105-012-0173-z
- Mandic A, Hansson J, Linder S, et al. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem 2003;278:9100–6. [PubMed] DOI:10.1074/jbc.M210284200
- Harhaji-Trajkovic L, Vilimanovich U, Kravic-Stevovic T, et al. AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumor cells. J Cell Mol Med 2009;13:3644–54. [PubMed] DOI:10.1111/j.1582-4934.2009.00663.x
- Cooley ME, Davis L, Abrahm J. Cisplatin: a clinical review. Part II-Nursing assessment and management of side effects of cisplatin. Cancer Nurs 1994;17:283–93.
- Bragodo P, Armesilla A, Silva A, et al. Apoptosis by cisplatin requires p53 mediated p38alpha MAPK activation through ROS generation. Apoptosis 2007;12:1733–42. [PubMed] DOI:10.1007/s10495-007-0082-8
- Fong MY, Jin S, Rane M, et al. Withaferin A synergizes the therapeutic effect of doxorubicin through ROS-mediated autophagy in ovarian cancer. PLoS One 2012;7:e42265. [PubMed] DOI:10.1371/journal.pone.0042265
- Stěrba M, Popelová O, Vávrová A, et al. Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid Redox Signal 2013;18:899–929. [PubMed] DOI:10.1089/ars.2012.4795
- De Larco JE, Park CA, Dronava H, et al. Paradoxical roles for antioxidants in tumor prevention and eradication. Cancer Biol Ther 2010;9:362–70. [PubMed] DOI:10.4161/cbt.9.5.10895
- Ozqur R, Turkan I, Uzilday B, et al. Endoplasmic reticulum stress triggers ROS signaling changes the redox state, and regulates the antioxidant defense of Arabidopsis thaliana. J Exp Bot 2014;65:1377–90. [PubMed] DOI:10.1093/jxb/eru034
- Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword. Antioxid Redox Signal 2007;9:2277–93. [PubMed] DOI:10.1089/ars.2007.1782
- Chen G, Izzo J, Demizu Y, et al. Different redox states in malignant and nonmalignant esophageal epithelial cells and differential cytotoxic responses to bile acid and honokiol. Antioxid Redox Signal 2009;11:1083–95. [PubMed] DOI:10.1089/ars.2008.2321
- Cai J, Jones DP. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem 1998;273:11401–4. [PubMed] DOI:10.1074/jbc.273.19.11401
- Hockenbery DM, Oltvai ZN, Yin XM, et al. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 1993;75:241–51. [PubMed] DOI:10.1016/0092-8674(93)80066-N
- Kane DJ, Sarafian TA, Anton R, et al. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science 1993;262:1274–7. [PubMed] DOI:10.1126/science.8235659
- Mayer M, Nobel M. N-acetyl-L-cysteine is a pluripotent protector against cell death and enhancer of troph as factor-mediated cell survival in vitro. Proc Natl Acad Sci USA 1994;91:7496–500. [PubMed] DOI:10.1073/pnas.91.16.7496
- Kluck RM, Bossy-Wetzel E, Green DR, et al. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 1997;275:1132–6. [PubMed] DOI:10.1126/science.275.5303.1132
- Jacobson MD, Raff MC. Programmed cell death and Bcl-2 protection in very low oxygen. Nature 1995;374:814–6. [PubMed] DOI:10.1038/374814a0
- Shimizu S, Eguchi Y, Kosaka H, et al. Prevention of hypoxia-induced cell death by Bcl-2 and Bcl-xL. Nature 1995;374:811–3. [PubMed] DOI:10.1038/374811a0
- Ko CH, Shen SC, Hsu CS, et al. Mitochondrial-dependent, reactive oxygen species-independent apoptosis by myricetin: role of protein kinase C, cytochrom c and caspase cascade. Biochem Pharmacol 2005;69:913–27. [PubMed] DOI:10.1016/j.bcp.2004.12.005
- Hou DX, Uto T, Tong X, et al. Involvement of reactive oxygen species-independent mitochondrial pathway in gossypol-induced apoptosis. Arch Biochem Biophys 2004;428:179–87. [PubMed] DOI:10.1016/j.abb.2004.06.007
- Lin S, Fujii M, Hou DX. Rhein induced apoptosis in HL-60 cells via reactive oxygen species-independent mitochondrial death pathway. Arch Biochem Biophys 2003;418:99–107. [PubMed] DOI:10.1016/j.abb.2003.08.004
- Carr AC, Zhu BZ, Frei B. Potential antiatherogenic mechanisms of ascorbate (vitamin C) and alpha-tocopherol (vitamin E). Circ Res 2000;87:349–54. [PubMed] DOI:10.1161/01.RES.87.5.349
- Frei B, Lawson S. Vitamin C and cancer revisited. Proc Natl Acad Sci USA 2008;105:11037–8. [PubMed] DOI:10.1073/pnas.0806433105
- Offord EA, Gautier JC, Avanti O, et al. Photoprotective potential of lycopene, beta-carotene, vitamin E, vitamin C and carnosic acid in UVA-irradiated human skin fibroblasts. Free Radic Biol Med 2002;32:1293–303. [PubMed] DOI:10.1016/S0891-5849(02)00831-6
- Stahl W, Sies H. Carotenoids and protection against solar UV radiation. Skin Pharmacol Appl Skin Physiol 2002;15:291–6. [PubMed] DOI:10.1159/000064532
- Trekli MC, Riss G, Goralczyk R, et al. Beta-carotene suppresses UVA-induced HO-1 gene expression in cultured FEK4. Free Radic Biol Med 2003;34:456–64. [PubMed] DOI:10.1016/S0891-5849(02)01303-5
- Black HS, Okotie-Eboh G, Gerguis J. Diet potentiates the UV-carcinogenic response to beta-carotene. Nutr Cancer 2000;37:173–8. [PubMed] DOI:10.1207/S15327914NC372_9
- Lin X, Zhang F, Bradbury CM, et al. 2-Deoxy-D-glucose-induced cytotoxicity and radiosensitization in tumor cells is mediated via disruptions in thiol metabolism. Cancer Res 2003;63:3413–7. [PubMed]
- Spitz DR, Sim JE, Ridnour LA, et al. Glucose deprivation-induced oxidative stress in human tumor cells. A fundamental defect in metabolism? Ann N Y Acad Sci 2000;899:349–62. [PubMed]
- Lin H, Hu YP, Savaraj N, et al. Hypersensitization of tumor cells to glucolytic inhibitors. Biochemistry 2001;40:5542–7. [PubMed] DOI:10.1021/bi002426w
- Lin H, Savaraj N, Priebe W, et al. Hypoxia increases tumor cell sensitivity to glucolytic inhibitors a strategy for solid tumor therapy (Model C). Biochem Pharmacol 2002;64:1745–51. [PubMed] DOI:10.1016/S0006-2952(02)01456-9
- Coleman MC, Asbury CR, Daniels D, et al. 2-deoxy-D-glucose causes cytotoxicity, oxidative stress, and radiosensitization in pancreatic cancer. Free Radic Biol Med 2008;44:322–31. [PubMed] DOI:10.1016/j.freeradbiomed.2007.08.032
- Aykin-Burns N, Ahmad IM, Zhu Y, et al. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J 2009;418:29–37. [PubMed] DOI:10.1042/BJ20081258
- Ding WQ, Vaught JL, Yamauchi H, et al. Differential sensitivity of cancer cells to docosahexaenoic acid-induced cytotoxicity: the potential importance of down-regulation of superoxide dismutase 1 expression. Mol Cancer Ther 2004;3:1109–17. [PubMed] DOI:10.4161/cbt.3.11.1190
- Zhelev Z, Ivanova D, Aoki I, et al. 2-deoxy-D-glucose sensitizes cancer cells to Barasertib and Everolimus by ROS-independent mechanism(s). Anticancer Res 2015;35:6623–32. [PubMed]
- Anel A, Naval J, Desportes P, et al. Increased cytotoxicity of polyunsaturated fatty acids on human tumoral B and T-cell lines compared with normal lymphocytes. Leukemia 1992;6:680–8. [PubMed]
- Heimli H, Hollung K, Drevon CA. Eicosapentaenoic acid-induced apoptosis depends on acyl CoA-synthetase. Lipids 2003;38:263–8. [PubMed] DOI:10.1007/s11745-003-1059-z
- Gleissman H, Johnsen JI, Kogner P. Omega-3 fatty acids in cancer, the protectors of good and the killers of evil. Exp Cell Res 2010;316:1365–73. [PubMed] DOI:10.1016/j.yexcr.2010.02.039
- Tuller ER, Beavers CT, Lou JR, et al. Docosahexaenoic acid inhibits superoxide dismutase 1 gene transcription in human cancer cells: the involvement of peroxisome proliferator-activated receptor alpha and hypoxia-inducible factor-2alpha signaling. Mol Pharmacol 2009;76:588–95. [PubMed] DOI:10.1124/mol.109.057430
- Huang H, Starodub O, Mclntosh A, et al. Liver fatty acid-binding protein targets fatty acids to the nucleus. Real time confocal and multiphoton fluorescence imaging in living cells. J Biol Chem 2002;277:29139–51. [PubMed] DOI:10.1074/jbc.M202923200
- Xia S, Lu Y, Wang J, et al. Melanoma growth is reduced in fat-1 transgenic mice: impact of omega-6/omega-3 essential fatty acids. Proc Natl Acad Sci USA 2006;103:12499–504. [PubMed] DOI:10.1073/pnas.0605394103
- Jia Q, Lupton JR, Smith R, et al. Reduced colitis-associated colon cancer in Fat-1 (n-3 fatty acid desaturase) transgenic mice. Cancer Res 2008;68:3985–91. [PubMed] DOI:10.1158/0008-5472.CAN-07-6251
- Lu Y, Nie D, Witt WT, et al. Expression of the fat-1 gene diminishes prostate cancer growth in vivo through enhancing apoptosis and inhibiting GSK-3 beta phosphorylation. Mol Cancer Ther 2008;7:3203–11. [PubMed] DOI:10.1158/1535-7163.MCT-08-0494
- Mahéo K, Vibet S, Steghens JP, et al. Differential sensitization of cancer cells to doxorubicin by DHA: a role for lipoperoxidation. Free Radic Biol Med 2005;39:742–51. [PubMed] DOI:10.1016/j.freeradbiomed.2005.04.023
- Vibet S, Goupille C, Bougnoux P, et al. Sensitization by docosahexaenoic acid (DHA) of breast cancer cells to anthracyclines through loss of glutathione peroxidase (GPx1) response. Free Radic Biol Med 2008;44:1483–91. [PubMed] DOI:10.1016/j.freeradbiomed.2008.01.009
- Lindskog M, Gleissman H, Ponthan F, et al. Neuro-blastoma cell death in response to docosahexaenoic acid: sensitization to chemotherapy and arsenic-induced oxidative stress. Int J Cancer 2006;118:2584–93. [PubMed] DOI:10.1002/(ISSN)1097-0215
- Rudra PK, Krokan HE. Cell-specific enhancement of doxorubicin toxicity in human tumor cells by docosahexaenoic acid. Anticancer Res 2001;21:29–38. [PubMed]
- Lin H, Hou CC, Cheng CF, et al. Peroxisomal proliferator-activated receptor-alpha protects rental tubular cells from doxorubicin-induced apoptosis. Mol Pharmacol 2007;72:1238–45. [PubMed] DOI:10.1124/mol.107.037523
- Zhelev Z, Ivanova D, Lazarova D, et al. Docosahexaenoic acid sensitizes leukemia lymphocytes to baraserib and everolimus by ROS-dependent mechanism without affecting the level of ROS and viability of normal lymphocytes. Anticancer Res 2016;36:1673–82. [PubMed]