
Endostatin enhances antitumor effect of tumor antigen-pulsed dendritic cell therapy in mouse xenograft model of lung carcinoma
Abstract
Objective: To investigate the antitumor effect of endostatin combined with tumor antigen-pulsed dendritic cell (DC)-T cell therapy on lung cancer.
Methods: Transplanted Lewis lung cancer (LLC) models of C57BL/6 mice were established by subcutaneous injection of LLC cells in left extremity axillary. Tumor antigen-pulsed DC-T cells from spleen cells and bone of mice were cultured in vitro. Tumor-bearing mice were randomly divided into three groups, including DC-T+endostatin group, DC-T group, and phosphate-buffered saline (PBS) control group. Microvessel density (MVD) of tumor tissue in tumor-bearing mice was determined by immunohistochemistry (IHC). The expressions of vascular endothelial growth factor (VEGF) and hypoxia-inducible factor-1α (HIF-1α) were determined by Western blotting and IHC staining. The proportions of CD8+ T cells, mature dendritic cells (mDC), tumor-associated macrophages [TAM (M1/M2)], and myeloid-derived suppressor cells (MDSC) in suspended cells of tumor tissue were determined by flow cytometry. The expressions of interleukin (IL)-6, IL-10, IL-17, transforming growth factor-β (TGF-β) and interferon-γ (IFN-γ) in suspended cells of tumor tissue were detected by enzyme-linked immune sorbent assay (ELISA).
Results: DC-T cells combined with endostatin remarkably suppressed tumor growth. MVD of mice in DC-T+endostatin group was significantly lower than that of the control group and DC-T monotherapy group. The expressions of VEGF, IL-6 and IL-17 in tumors were markedly decreased, but IFN-γ and HIF-1α increased after treating with DC-T cells combined with endostatin, compared to control group and DC-T group. In the DC-T+endostatin group, the proportions of MDSC and TAM (M2 type) were significantly decreased, mDC and TAM (M1 type) were up-regulated, and CD8+ T cells were recruited to infiltrate tumors, in contrast to PBS control and DC-T monotherapy. DC-T cells combined with endostatin potently reduced the expressions of IL-6, IL-10, TGF-β and IL-17 in tumor tissue, and enhanced the expression of IFN-γ.
Conclusions: The study indicated the synergic antitumor effects between endostatin and tumor antigen-pulsed DC-T cells, which may be a prospective therapy strategy to achieve potent antitumor effects on lung cancer.
Keywords: Endostatin; DC-T cells; lung cancer; cellular therapy; tumor microenvironment
Submitted Mar 29, 2016. Accepted for publication Jul 31, 2016.
doi: 10.21147/j.issn.1000-9604.2016.04.09
Introduction
In recent years, cancer has become a major public health problem worldwide (1). Traditional therapies like surgery, chemotherapy and radiotherapy are less effective for many cancer patients. Cellular immunotherapy is an effective method to treat some cancers that traditional therapies have failed to control (2, 3). However, its clinical benefit rate is relatively low in general (4). Recent studies of cancer biotherapy focus on developing a more targeted, effective and safer strategy (5-8). The molecular identification of human cancer-specific antigens has powerfully improved the development of antigen-specific immunotherapy. There are multiple sources and types of T cells used for adoptive therapy, including expanded and activated tumor infiltrating lymphocytes, T cells modified ex vivo to express a specific T cell receptor, and cytotoxic T cells sensitized in vitro by tumor antigen-pulsed dendritic cells (DC)-T cells (9-12).
Inflammatory microenvironment is one of the hallmarks of nearly all tumors (13-15). Immunocytes [e.g., myeloid-derived suppressor cells (MDSC), tumor-associated macrophages (TAM) and immature dendritic cells (imDC)] and pro-inflammatory cytokines [e.g., interleukin (IL)-6, IL-17, tumor necrosis factor (TNF)-α and chemokines] in tumor microenvironment markedly facilitate tumor progression through the stimulation of neovascularization and tissue remodeling, and hence, leading to suppressive immune state (15, 16). It has been shown that tumor microenvironment plays a key role in the success of cellular immunotherapy by transforming adoptively transferred killer T cells into suppressive regulatory T cells with the help of several inhibitory factors like MDSC (17).
Angiogenesis is another basic biological characteristic of tumor (18). Targeting tumor angiogenesis is a therapeutic strategy with good prospects for the control of tumor growth (19, 20). Endostatin is a potential antiangiogenic agent that has scarcely any toxicity and drug resistance (21). It has been proved by several preclinical studies that synergy exists between vascular endothelial growth factor (VEGF)-targeted antiangiogenic agents and immunotherapy (22). Normalization of tumor vasculature caused by anti-VEGF antibody can increase the infiltration of adoptively transferred T cells into tumors and improve the effectiveness of adoptive cell transfer-based immunotherapy in tumor-bearing mouse models (23). However, the impact of endostatin on the antitumor effect of cellular immunotherapy is not clear and the synergy of immunotherapy with endostatin is urgently needed to be investigated. In this study, we explored the influence of endostatin on the antitumor effect of tumor antigen-pulsed DC-T cells, in order to provide a prospective therapy strategy to achieve potent antitumor effect by combining endostatin with cellular immunotherapy.
Materials and methods
Cells
Lewis lung cancer (LLC) cell line (from lung adeno-carcinoma cell line of C57BL/6 mice) was purchased from Shanghai Cell Bank of Chinese Academy of Sciences and was cultured in Dulbecco's modified eagle medium (DMEM) containing 10% fetal bovine serum (FBS). Cells (1×107) were resuspended in RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) and mixed. They were then repeatedly frozen and thawed at -80 ℃ and 42 ℃ for three times. After cell disruption, the cells were centrifuged at 15, 000 r/min for 30 min. The supernatant was then collected, filtered, sterilized and stored at 4 ℃.
Antibodies and reagents
Recombinant human endostatin (rhEndostatin, Simcere Pharm, Nanjing, China); EZ-SepTM Mouse percollase (Amresco); RPMI 1640 medium, FBS (GIBCO); ConA, DMEM medium, phosphate-buffered saline (PBS) buffer (SIGMA); fluorescently-labeled antibody CD3, CD4, CD8, CD11c, CD86, major histocompatibility complex (MHC) II, CD11b, Gr-1, CD206, CD68 and NOS2 and their isotype controls (eBioscience); Mouse Lymphocyte Factor ELISA Kit (Shanghai Enzyme-linked Biotechnology Co., Ltd.); BCA Protein Assay Kit (Beyotime); anti-mouse hypoxia-inducible factor-1α (HIF-1α), VEGF antibody (Abcam); rmGM-CSF, rmIL-4 (Peprotech), rmIL-2, rmTNF-α (BIOLOGICAL); anti-mouse CD31 non-labeled immunohistochemical monoclonal antibody (Santa Cruz); MCO-15AC CO2 incubator (SANYO); sterile 1.5 laminar flow bechtop (Thermo Scientific); FACS Calibur flow cytometer (Becton Dickinson); NanoDrop ND-1000 ultraviolet spectrophotometer (Agilent); Model 680 Microplate Reader (Bio-Rad).
Animals
Male wild-type C57BL/6 mice (age, 6 weeks; weight, 18-22 g) were purchased from Beijing Laboratory Animal Center of Chinese Academy of Sciences and fed in a specific-pathogen-free animal laboratory. The feeding and use of laboratory animals complied with Animal Experimentation Ethical Standards proposed by Ethics Committee of Shandong University [SCXK (Lu) 2003-0003].
After LLC cells were recovered and subcultured in complete medium, the cells in log phase were used and cell concentration was adjusted to 1×107/mL. Right rib skin of C57BL/6 mice was disinfected with 75% alcohol and suspension of LLC cells was collected with 1 mL syringe (mixing upside down). Suspension (0.2 mL) was then given to each mouse via subcutaneous injection, with 1×106 cells being inoculated in each mouse.
Tumor antigen-pulsed DC-T cells
The bilateral femur and tibia of a mouse were separated under aseptic condition and both ends of the bones were cut off. Then we took out RPMI 1640 medium with a 1 mL syringe and inserted the medium into marrow cavity from both ends of the bones. Bone marrow was therefore flushed into a culture plate. We repeated this step 4-6 times until the marrow cavity became white. A sterile glass rod was used to grind the marrow. Marrow suspension was collected and murine lymphocyte separation medium was added into it. The obtained solution was centrifuged. Buffy coat in the upper and middle layers was carefully collected along the tube wall to obtain cells (mainly mononuclear cells). Then, the obtained cells were transferred into RPMI 1640 complete medium which was more than 5 times the volume of the cells. The cells and the medium were mixed, centrifuged and washed twice to collect cells. Cells were put into the incubator with 5% CO2 and incubated at 37 ℃ for 48 h after cell concentration was adjusted to 1×106/mL. Adherent cells were kept and added into complete medium. Medium was half exchanged every other day and cytokine was replenished. On d 6 after culture, DC were co-cultured with LLC cell frozen-thawed antigen at 1:5 with rmTNF-α 20 ng/mL added. The obtained solution was placed into the incubator with 5% CO2 and incubated at 37 ℃. We observed cell colony and recorded DC growth under inverted microscope. On d 9, by gently blowing and beating, we collected all suspension cells which were mouse myeloid mature dendritic cells (mDC). The collected mDC were then tested.
After separation and on d 9 after culture of DC and T cells, mDC were collected and added into mitomycin solution (with concentration adjusted at 25 μg/mL). The solution was then incubated at 37 ℃ for 30 min. DC were mixed with splenic lymphocytes at 1:10. The mixture was then added into an incubator with 5% CO2 and incubated at 37 ℃ for 24 h. The obtained cells were tumor antigen-specific DC-T cells.
In vivo experiments design
C57BL/6 mice were divided into three groups (DC-T alone, DC-T+endostatin and control) with 7 mice in each group. Intervention was given to tumor-bearing mice on d 7. We observed tumor growth and measured tumor diameter every other day. The mice were killed 24 h after administration on d 14. In control group, PBS (0.2 mL) was given to each C57BL/6 mouse daily by intravenous injection via tail for a total of 14 d. In DC-T+endostatin group, rhEndostatin was given to each mouse by tail intravenous injection for a total of 14 d at a dose of 15 mg/kg daily. In DC-T group and DC-T+endostatin group, 5×106 tumor antigen-pulsed DC-T cells were given to each mouse by tail intravenous injection on d 7 after the model of tumor-bearing C57BL/6 mice was established.
To measure mouse weight and tumor inhibition rate, electronic scale was used to measure the weight of mice in each group every other day. After tumor-bearing C57BL/6 mice were killed, tumor tissues were dissected and weighed. Tumor inhibition rate = (1-mean tumor weight of treatment groups/mean tumor weight of control group) ×100%. For the measurement of tumor volume, vernier caliper was used to measure the longest and shortest diameter of tumors. Then, mean tumor volume and tumor inhibition rate in each group were calculated and growth curve was drawn. Tumor volume = long diameter of tumor × short diameter of tumor2/2.
Microscopy
Tumor tissues were separated from tumor-bearing mice and fixed with paraformaldehyde. The tissues were embedded with paraffin and then sectioned. The paraffin sections were dewaxed to water. Streptavidin-peroxidase method was used to repair antigen under high pressure for 8 min. Subsequent procedures were conducted according to instructions of secondary antibody kit and DAB Color Development Kit (Bio-Rad, Hercules, CA, USA). Under a light microscope, mean microvessel density (MVD) was counted in 6 high-power fields (200×) for each section.
Flow cytometry
Tumor tissues were dissected and cut into pieces. After trypsinization, the tissues were filtered through 300 molybdenum sieving mesh to obtain monocytes. After cell density was adjusted to 5×105/mL, 100 μL cell suspension was added into each flow tube. Flow tubes contained phycoerythrin-labeled anti-CD83 and anti-CD86 antibodies (BioLegend, San Diego, CA, USA), allophycocyanin-labeled anti-CD68 antibody, phycoerythrin-labeled anti-iNOS antiboody, fluorescein isothiocyanate-labeled anti-CD3, anti-CD4, anti-interferon-γ (IFN-γ), anti-CD206, anti-Gr-1 and anti-CD11c antibodies (eBioscience, San Diego, CA, USA). Negative control, isotype control and single line pipette groups were designed. Phycoerythrin-rat immunoglobulin (Ig) G and fluorescein isothiocyanate-hamster IgG were added in control groups. After being fully mixed with fluorescent-labeled antibody, they were placed at room temperature away from light for 15 min. FACS Aria II sorting flow cytometer (BD Biosciences, San Jose, CA, USA) was used to detect molecule expression on cell surface and CellQuest software was used to analyze data.
Enzyme-linked immune sorbent assay (ELISA)
Antibodies were diluted with coating buffer to the extent where protein level was 1-10 µg/mL. Then, 0.1 mL of antibodies were taken and added to ELISA plate wells, which were incubated at 4 ℃ overnight. The required number of wells was calculated according to the number of test samples, blank control and standard samples. Distilled water (130 μL) was added into blank and standard wells, while 100 μL cell supernatant was added into sample wells for testing. Each sample group was made in duplicate or triplicate. The plate was covered and incubated in an incubator with 5% CO2 at 37 ℃ for 90 min, afterwards, liquid in wells was discarded and the wells were washed 3 times. After 100 μL biotin antibodies were added to each well, the plate was covered and incubated in an incubator with 5% CO2 at 37 ℃ for 1 h. Then, the plate was washed with washing liquid for 3 times of 3 min. Enzyme conjugate (100 μL) was added into each well and the plate was incubated in the incubator with 5% CO2 at 37 ℃ for 30 min. Afterwards, the plate was washed for 3 times. Color developing reagent (100 μL) was added into each well and kept away from light. The plate was incubated in the incubator with 5% CO2 at 37 ℃ for 10-20 min. Finally, 100 μL stop buffer was added into each well to terminate reaction and OD450 values were determined using a microplate reader (Model 680; Bio-Rad, Hercules, CA, USA).
Western blotting
After tumor tissues were taken out from tumor-bearing mice. They were chipped and then ground in a homogenizer. Total protein was extracted through disruption on ice, and 400 μL RIPA lysis buffer was added to each group. After disruption, cells were taken out and transferred to a centrifugal tube of 1.5 mL. Then, cells were centrifuged at 12, 000 r/min at 4 ℃ for 20 min, and the supernatant was collected. BCA Protein Assay Kit was used to determine the concentration of extracted protein (Beyotime, Shanghai, China). Sodium dodecyl sulfate polyacrylamide gel electrophoresis was used to transfer protein to polyvinylidene fluoride film. The film was then covered with Tris-buffered saline and Tween 20 (TBST) containing 5% skimmed milk for 2 h. After primary antibodies (rabbit anti-mouse HIF-1α monoclonal antibody, 1:1, 000; rabbit anti-mouse VEGF monoclonal antibody, 1:1, 000; Abcam, Cambridge, USA) were added, the film was incubated at 4 ℃ overnight. The film was washed 5 times (6 min each) with TBST. After secondary antibody (horseradish peroxidase goat anti-mouse IgG antibody conjugate, 1:2, 000; Abcam, Cambridge, USA) was added, the film was incubated at 37 ℃ for 1 h and washed with TBST. Electrochemiluminescence kit was used to achieve chemiluminescence signals. SmartView electrophoresis image analysis system (Smartview Enterprise Imaging Solutions, Irvine, CA, USA) was used to obtain images. Quantity One software (Bio-Rad, Hercules, CA, USA) was applied to analyze gray values of each zone. The ratio of gray values of each interested protein to β-actin was calculated for statistical analysis.
Immunohistochemisty (IHC)
Tumor tissues were separated from tumor-bearing mice and fixed with formalin. The tissues were embedded with paraffin and then sectioned. The paraffin sections were dewaxed to water. Streptavidin-peroxidase method was used following kit instructions. Primary antibodies were replaced with PBS as negative control. According to semi-quantitative integration, the images were reviewed by two physicians from the Department of Pathology and a conclusion was drawn. The results were scored according to positive staining intensity and expression of positive cells. Positive staining intensity: cells had no staining (score 0); light brown cells were weakly positive (score 1); brown cells were moderately positive (score 2); and brown cells without background coloring were strongly positive (score 3). For the expression of positive cells, 5 different fields were chosen in 400× light microscope and 200 cells were counted for each field. The percentage of positive cells was then calculated. The score was 0 if positive cells ≤5%; 1 if positive cells ≤25%; 2 if 25%< positive cells ≤50%; and 3 if positive cells >50%. There were four grades of IHC results according the above scores: 0 was rated as negative (-), 1-4 were weakly positive (+), 5-8 were moderately positive (+), and 9-12 were strongly positive (+++).
Statistical analysis
All data were analyzed using SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). Measurement data were expressed as x±s. Statistical differences between groups were determined using one-way analysis of variance (ANOVA), and LSD test was performed to make comparison between any two groups. Quantitative comparison was conducted by χ2 test and Fisher's exact test. α=0.05 and P<0.05 were considered statistically significant.
Results
DC-T+endostatin strongly inhibit tumor angiogenesis and significantly reduce tumor growth
Tumor antigen-pulsed DC-T cells actively suppressed tumor growth compared with control group (P=0.021), while tumor antigen-pulsed DC-T cells combined with endostatin were more effective in inhibiting tumor growth compared with control group (P=0.009) (Figure 1A, 1B). Using CD31 as the marker of vascular endothelial cells, cytoplasm of endothelial cells was yellowish-brown after IHC staining. Tumor MVD was less in mice receiving tumor antigen-pulsed DC-T cells compared with control group (P=0.027), and MVD was significantly decreased in mice receiving tumor antigen-pulsed DC-T cells combined with endostatin compared with control group (P=0.002), with a marked increase in necrosis of tumor tissues (Figure 1C, 1D). These results indicate that tumor antigen-pulsed DC-T cells combined with endostatin markedly decrease tumor vasculature, strongly inhibit tumor angiogenesis and significantly reduce tumor growth compared with DC-T cells alone or PBS control.
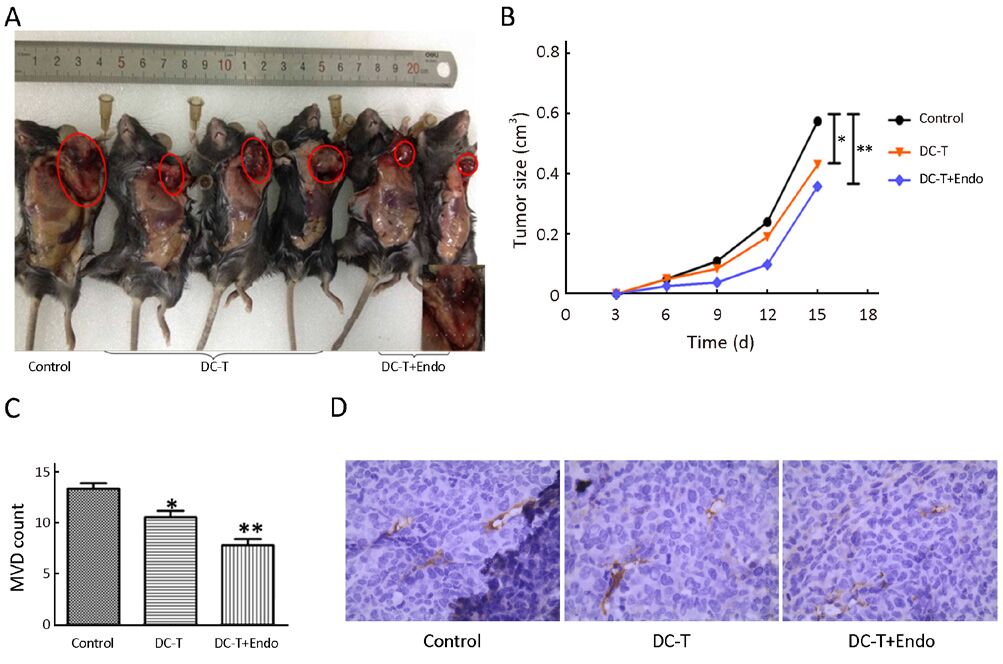
DC-T+endostatin down-regulate multiple proangiogenic factors and augment hypoxia in tumors
According to Western blotting results, VEGF expression in tumors was markedly reduced in DC-T alone group compared with PBS control (P=0.002), while the expression of HIF-1α in DC-T alone group was higher than that in PBS control group (P=0.000). In addition, VEGF expression was significantly reduced (P=0.000) and HIF-1α expression was markedly increased (P=0.000) in the DC-T+endostatin group compared with control (Figure 2A, 2B). These results were supported by the outcomes of IHC staining (Table 1, 2; Figure 2C). ELISA showed that tumor antigen-pulsed DC-T cells significantly decreased the expression of IL-6 and IL-17 (P=0.026 and 0.044, respectively), but increased the expression of IFN-γ (P=0.026). Of note, the levels of IL-6 and IL-17 were significantly reduced (P=0.008 and 0.010, respectively) and IFN-γ expression was markedly increased (P=0.009) in the DC-T+endostatin group (Figure 2D). These results suggest that the combination of tumor antigen-specific DC-T cells and endostatin significantly inhibits the expression of proangiogenic factors and increases the expression of anti-angiogenic factors, while markedly increasing HIF-1α expression and intensifying hypoxia in tumors.
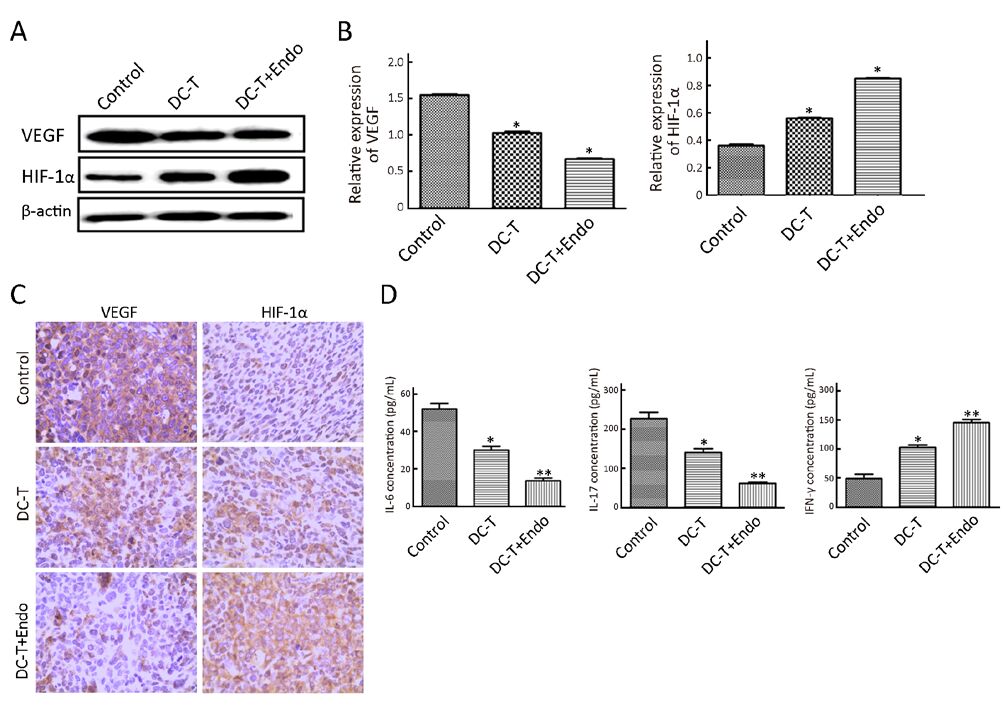
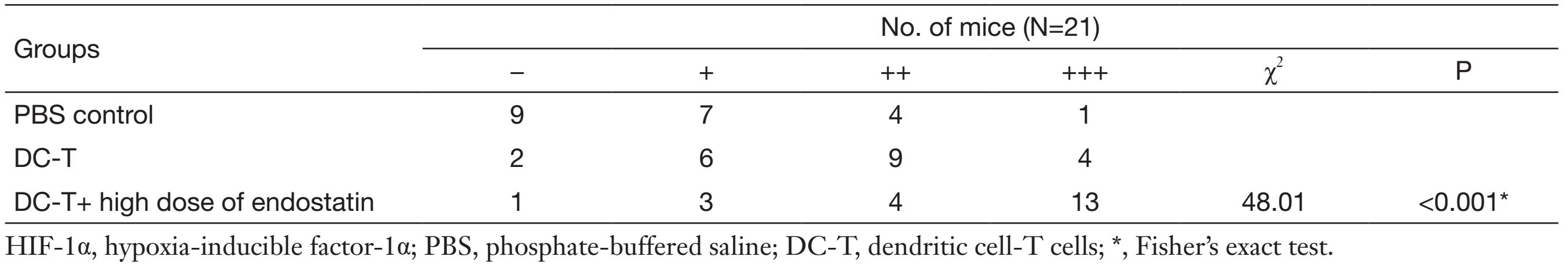
Full table
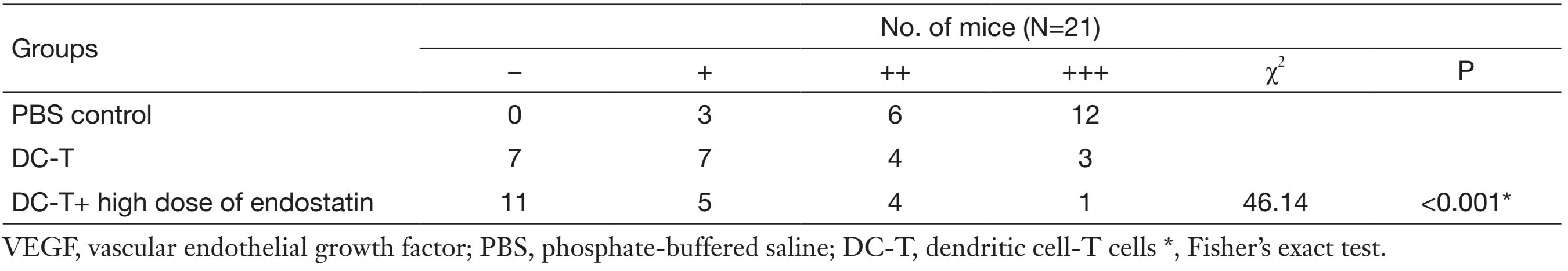
Full table
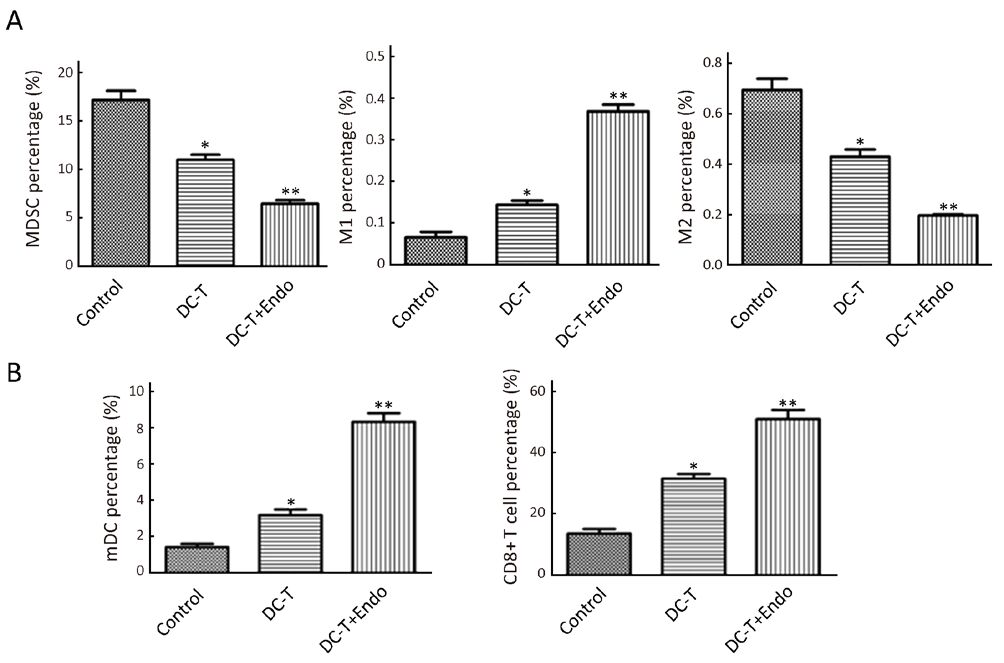
DC-T+endostatin significantly decrease immunosuppressive cells in tumor tissues and increase tumor-infiltrated mDC and CD8+ T cells
As suggested by flow cytometry, immunosuppressive MDSC was reduced (P=0.030), M1 TAM with immunological enhancement was increased (P=0.045) and M2 TAM with immunosuppression was decreased (P=0.038) in DC-T alone group. Moreover, tumor antigen-pulsed DC-T cells in combination with endostatin significantly decreased MDSC in tumors (P=0.009), markedly increased M1 TAM (P=0.005) and significantly decreased M2 TAM (P=0.008) compared with PBS control (Figure 3A). Tumor-infiltrated mDC (P=0.038) and CD8+ T (P=0.019) cells were increased in DC-T alone group compared with PBS control. Tumor antigen-pulsed DC-T cells in combination with endostatin significantly up-regulated mDC (P=0.005) and increased tumor-infiltrated CD8+ T cells (P=0.008) (Figure 3B). These results indicate that, compared with DC-T cells alone and PBS control, tumor antigen-specific DC-T cells in combination with endostatin significantly decrease immunosuppressive MDSC and M2 TAM, increase M1 TAM and elevate tumor-infiltrated mDC and CD8+ T cells, effectively reversing immunosuppression in tumor microenvironment.
DC-T+endostatin down-regulate expression of immunoinhibitory cytokines in tumors and increase expression of IFN-γ involved in oncolysis
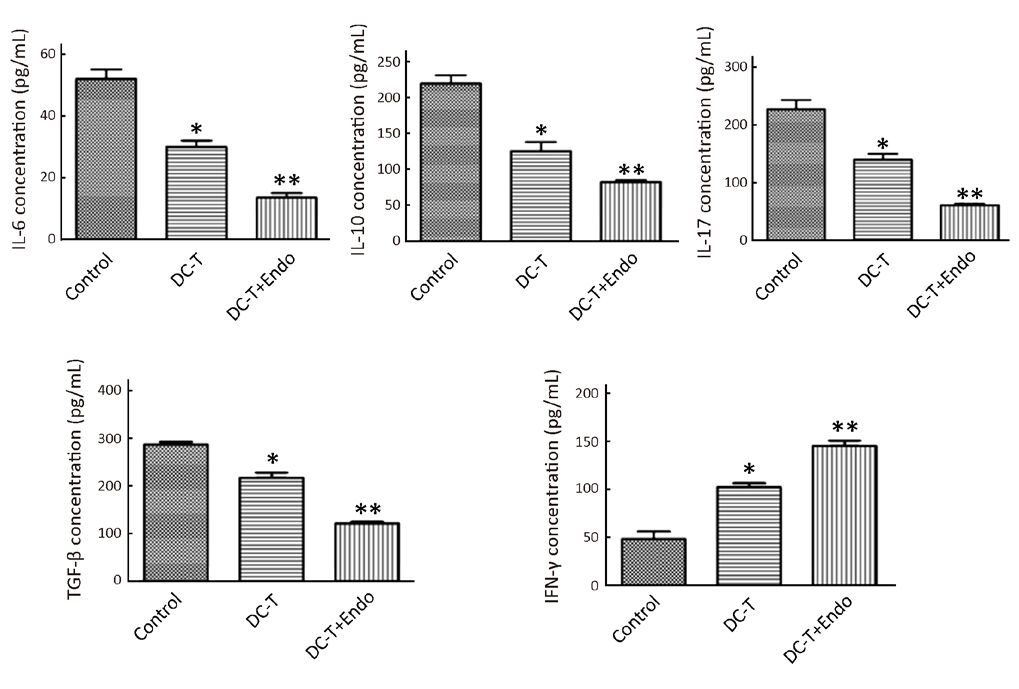
As shown by ELISA, DC-T cells alone down-regulated immunoinhibitory cytokines [IL-6, IL-10, transforming growth factor-β (TGF-β) and IL-17] (P=0.026, 0.032, 0.029 and 0.044, respectively) and increased the expression of IFN-γ involved in oncolysis (P=0.026), compared with PBS control. Furthermore, tumor antigen-pulsed DC-T cells combined with endostatin significantly down-regulated the expression of IL-6, IL-10, TGF-β and IL-17 (P=0.008, 0.008, 0.002 and 0.010, respectively) and markedly increased the expression of IFN-γ (P=0.009) (Figure 4). These results suggest that the combination of DC-T cells and endostatin down-regulates multiple immunoinhibitory cytokines and the expression of related cytokines, up-regulates M1 TAM, increases the expression of IFN-γ involved in oncolysis and facilitates the infiltration of mDC and CD8+ T cells into tumor tissues.
Discussion
With the understanding of the close connection between tumor progression and immunoescape, the pivotal role of immune-effector cells in oncobiology and the treatment of tumor have attracted much attention from researchers. Cellular immunotherapy has made considerable contributions to tumor therapy by effectively treating some cancers that chemotherapy has failed to control (3, 24). Despite inspiring advances, the clinical benefit rate of cellular immunotherapy is relatively low. There is a big difference between significant antitumor effect shown by preclinical researches and lower clinical benefit rates (2-4). Recent studies on cancer biotherapy are focused on discovering a new mode of combination cancer therapy that is targeted, safe and effective to improve the efficacy of cellular immunotherapy in treatment of cancers (5-8).
It has been shown that, although adoptively transferred T cells are cancer-specific and gather in tumor mass, tumor microenvironment plays a decisive role in the success of cellular immunotherapy by transforming adoptively transferred killer T cells into suppressive regulatory T cells by virtue of several inhibitory factors (17). VEGF antagonists (e.g., bevacizumab) or small-molecule inhibitors (e.g., sunitinib) are conducive to the formation of potent antitumor T cells by reversing the above-mentioned process (23, 25-27). Several reports have shown that the number of vaccine-induced T cells infiltrated into tumors can be increased by inhibiting VEGF activity (28-32). Normalization of tumor vasculature caused by anti-VEGF antibody can increase the infiltration of adoptively transferred T cells into tumors and improve the effectiveness of adoptive cellular immunotherapy in tumor-bearing mouse models (23, 33). These preclinical studies indicate that there is synergy between VEGF-targeted antiangiogenic agents and immunotherapy (28, 30, 34). However, these findings mainly come from studies on anti-VEGF monoclonal antibody or tyrosine kinase inhibitor (sunitinib). It is important to note that the effect of certain antiangiogenic molecules cannot be generalized to all antiangiogenic agents according to preliminary results (35, 36). This indicates that it is controversial whether VEGF should be the main target of antiangiogenesis and immunosuppression reversion. In particular, most antiangiogenic molecules inhibit not only VEGF signaling pathway, but also alternative pathways that may activate immune cells or factors (e.g., platelet-derived growth factor, c-Kit, fit-3). However, the impact of endostatin on the antitumor effect of cellular immunotherapy is not clear and it remains to be determined whether the synergy between antiangiogenic agents and cellular immunotherapy can be generalized to endostatin.
In this study, we used tumor antigen-pulsed DC-T cells in combination with endostatin to treat tumor-bearing mice with LLC. Compared with control and DC-T cells alone, adoptively transferred tumor antigen-specific DC-T cells combined with endostatin decreased the expression of proangiogenic factors (VEGF, IL-6 and IL-17) in the tumor microenvironment, strongly inhibited tumor angiogenesis, augmented hypoxia in tumor tissues and significantly suppressed tumor growth. In addition, it also reduced immunosuppressive MDSC and M2 TAM, down-regulated immunoinhibitory cytokines (IL-6, IL-10, TGF-β and IL-17), increased M1 TAM and the expression of IFN-γ involved in oncolysis and significantly elevated tumor-infiltrated mDC and CD8+ T cells so as to reverse immunosuppression in tumor microenvironment and enhance antitumor effect.
Although the efficacy of antiangiogenic therapy has been demonstrated by clinical practice, long-term clinical benefit of antiangiogenic agents has not been obtained in most cancer patients (33, 37). As many pathways affected by antitumor targeted molecular therapy also play critical roles in immune function, it is possible to achieve the best antitumor immune effect of immunotherapy by virtue of targeted therapy (28). Theoretically, clinical response characterized by suppressive effect may be transformed to long-term effect of tumor regression by targeted therapy with the assistance of immunotherapy, while targeted therapy may weaken tumor-mediated immunosuppression by eliminating inflammatory reaction caused by tumors and decreasing immunoinhibitory cells (38). In our study, we demonstrated that endostatin, an antiangiogenic agent, in combination with tumor antigen-pulsed DC-T cells, may significantly improve antitumor effect by potently inhibiting tumor angiogenesis, reversing immunosuppression in tumor microenvironment and increasing infiltration of killer T cells into tumor tissues.
Conclusions
The study indicated the synergic antitumor effects between endostatin and tumor antigen-pulsed DC-T cells. These findings may serve as a theoretical and experimental foundation for combining antiangiogenic therapy of endostatin with cellular immunotherapy to achieve potent antitumor effects.
Acknowledgements
Funding: This work was supported by Natural Science Foundat ion of Shandong province, China (No. ZR2010HL015), and Natural Science Youth Foundation of Shandong province, China (No. ZR2013HQ017).
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30. [PubMed] DOI:10.3322/caac.v66.1
- Sharma P, Wagner K, Wolchok JD, et al. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer 2011;11:805–12. [PubMed] DOI:10.1038/nrc3153
- McNutt M. Cancer immunotherapy. Science 2013;342:1417. [PubMed] DOI:10.1126/science.1249481
- Rosenberg SA. Overcoming obstacles to the effective immunotherapy of human cancer. Proc Natl Acad Sci USA 2008;105:12643–4. [PubMed] DOI:10.1073/pnas.0806877105
- Pao W. New approaches to targeted therapy in lung cancer. Proc Am Thorac Soc 2012;9:72–3. [PubMed] DOI:10.1513/pats.201112-054MS
- Blank CU. The perspective of immunotherapy: new molecules and new mechanisms of action in immune modulation. Curr Opin Oncol 2014;26:204–14. [PubMed] DOI:10.1097/CCO.0000000000000054
-
Romagnoli GG, Zelante BB, Toniolo PA, et al. Dendritic
cell-derived exosomes may be a tool for cancer immunotherapy by converting tumor cells into immunogenic targets. Front Immunol 2015;5:692. [PubMed] - Sabado RL, Bhardwaj N. Dendritic cell immunotherapy. Ann N Y Acad Sci 2013;1284:31–45. [PubMed] DOI:10.1111/nyas.2013.1284.issue-1
- H Yi D, Appel S. Current status and future perspectives of dendritic cell-based cancer immunotherapy. Scand J Immunol 2013;78:167–71. [PubMed] DOI:10.1111/sji.2013.78.issue-2
- Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012;12:265–77. [PubMed] DOI:10.1038/nrc3258
- Wang K, Zhou Q, Guo AL, et al. An autologous therapeutic dendritic cell vaccine transfected with total lung carcinoma RNA stimulates cytotoxic T lymphocyte responses against non-small cell lung cancer. Immunol Invest 2009;38:665–80. [PubMed] DOI:10.1080/08820130903070528
- Yang S, Archer GE, Flores CE, et al. A cytokine cocktail directly modulates the phenotype of DC-enriched anti-tumor T cells to convey potent anti-tumor activities in a murine model. Cancer Immunol Immunother 2013;62:1649–62. [PubMed] DOI:10.1007/s00262-013-1464-0
- Giakoustidis A, Mudan S, Hagemann T. Tumour microen-vironment: overview with an emphasis on the colorectal liver metastasis pathway. Cancer Microenviron 2015;8:177–86. [PubMed] DOI:10.1007/s12307-014-0155-5
- Hattar K, Franz K, Ludwig M, et al. Interactions between neutrophils and non-small cell lung cancer cells: enhancement of tumor proliferation and inflammatory mediator synthesis. Cancer Immunol Immunother 2014;63:1297–306. [PubMed] DOI:10.1007/s00262-014-1606-z
- Grivennikov SI, Greten FR, Karin M. Immunity, inflam-mation, and cancer. Cell 2010;140:883–99. [PubMed] DOI:10.1016/j.cell.2010.01.025
- Keibel A, Singh V, Sharma MC. Inflammation, micro-environment, and the immune system in cancer progression. Curr Pharm Des 2009;15:1949–55. [PubMed] DOI:10.2174/138161209788453167
- Vasievich EA, Huang L. The suppressive tumor micro-environment: a challenge in cancer immunotherapy. Mol Pharm 2011;8:635–41. [PubMed] DOI:10.1021/mp1004228
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [PubMed] DOI:10.1016/j.cell.2011.02.013
- Bruno A, Pagani A, Magnani E, et al. Inflammatory angiogenesis and the tumor microenvironment as targets for cancer therapy and prevention. Cancer Treat Res 2014;159:401–26. [PubMed] DOI:10.1007/978-3-642-38007-5
- Samples J, Willis M, Klauber-deMore N. Targeting angiogenesis and the tumor microenvironment. Surg Oncol Clin N Am 2013;22:629–39. [PubMed] DOI:10.1016/j.soc.2013.06.002
- Folkman J. Antiangiogenesis in cancer therapy-endostatin and its mechanisms of action. Exp Cell Res 2006;312:594–607. [PubMed] DOI:10.1016/j.yexcr.2005.11.015
- Welti J, Loges S, Dimmeler S, et al. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest 2013;123:3190–200. [PubMed] DOI:10.1172/JCI70212
- Shrimali RK, Yu Z, Theoret MR, et al. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res 2010;70:6171–80. [PubMed] DOI:10.1158/0008-5472.CAN-10-0153
- Sonpavde G, Agarwal N, Choueiri TK, et al. Recent advances in immunotherapy for the treatment of prostate cancer. Expert Opin Biol Ther 2011;11:997–1009. [PubMed] DOI:10.1517/14712598.2011.575357
- Ko JS, Zea AH, Rini BI, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res 2009;15:2148–57. [PubMed] DOI:10.1158/1078-0432.CCR-08-1332
- Ozao-Choy J, Ma G, Kao J, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res 2009;69:2514–22. [PubMed] DOI:10.1158/0008-5472.CAN-08-4709
- Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer--what clinicians need to know. Nat Rev Clin Oncol 2011;8:577–85. [PubMed] DOI:10.1038/nrclinonc.2011.116
- Griffioen AW. Anti-angiogenesis: making the tumor vulnerable to the immune system. Cancer Immunol Immunother 2008;57:1553–8. [PubMed] DOI:10.1007/s00262-008-0524-3
- Johnson BF, Clay TM, Hobeika AC, et al. Vascular endothelial growth factor and immunosuppression in cancer: current knowledge and potential for new therapy. Expert Opin Biol Ther 2007;7:449–60. [PubMed] DOI:10.1517/14712598.7.4.449
- Huang Y, Yuan J, Righi E, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci USA 2012;109:17561–6. [PubMed] DOI:10.1073/pnas.1215397109
- Chan SF, Wang HT, Huang KW, et al. Anti-angiogenic therapy renders large tumors vulnerable to immunotherapy via reducing immunosuppression in the tumor micro-environment. Cancer Lett 2012;320:23–30. [PubMed] DOI:10.1016/j.canlet.2012.01.024
- Osada T, Chong G, Tansik R, et al. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol Immunother 2008;57:1115–24. [PubMed] DOI:10.1007/s00262-007-0441-x
- Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell 2011;146:873–87. [PubMed] DOI:10.1016/j.cell.2011.08.039
- Schoenfeld JD, Dranoff G. Anti-angiogenesis immuno-therapy. Hum Vaccin 2011;7:976–81. [PubMed] DOI:10.4161/hv.7.9.16407
- Teicher BA. Antiangiogenic agents and targets: A perspec-tive. Biochem Pharmacol 2011;81:6–12. [PubMed] DOI:10.1016/j.bcp.2010.09.023
- Schoenfeld J, Jinushi M, Nakazaki Y, et al. Active immuno-therapy induces antibody responses that target tumor angiogenesis. Cancer Res 2010;70:10150–60. [PubMed] DOI:10.1158/0008-5472.CAN-10-1852
- Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol 2011;8:210–21. [PubMed] DOI:10.1038/nrclinonc.2011.21
- Nair S, Boczkowski D, Moeller B, et al. Synergy between tumor immunotherapy and antiangiogenic therapy. Blood 2003;102:964–71. [PubMed] DOI:10.1182/blood-2002-12-3738