
Differential DNA methylation profiles of human B lymphocytes and Epstein-Barr virus-immortalized B lymphocytes
Introduction
Epstein-Barr virus (EBV) is a tumorigenic human herpesvirus which promotes proliferation and inhibits apoptosis in infected cells (1). The causative association of EBV with cancer was initially established in Burkitt’s lymphoma by the finding that EBV infection could immortalize B cells in vitro, generating continuously proliferating lymphoblastoid cell lines (LCL) (2). Although several viral products were necessary to maintain B-cell proliferation and several genes targeted by these viral proteins had been described, EBV-induced immortalization was not well understood (2,3). Several studies suggested that epigenetic alterations of human genome by these viral proteins might play a critical role in the process of B-cell immortalization (2,4,5). However, the contribution of EBV to methylation alterations has not been fully elucidated. Comparison of DNA methylome in human primary B cells and matched LCLs is an appropriate model to adequately and comprehensively characterize EBV-induced methylation alterations of B cells, which could give a hint on the methylation mechanism of EBV-induced immortalization.
However, most previous studies characterized the effects of EBV infection on only a few selected genes (6,7), and recent genome-wide DNA methylation studies compared methylation variations between LCLs and white blood cells (WBCs) (8-11) or mainly focused on measuring DNA methylation alterations in promoter or CpG islands (4,12). Since methylation status varied considerably between B cells and other WBC types per se and additionally aberrant methylation changes in CpG island shores or gene bodies besides CpG islands or promoter also play a critical role in many diseases (13,14), it is necessary to comprehensively evaluate methylation alterations across whole genome between human primary B cells and matched LCLs.
To comprehensively assess EBV-induced methylation alterations of human B cells across whole genome, we performed genome-wide DNA methylation analysis based on the Infinium MethylationEPIC BeadChip (Illumina, San Diego, CA, USA) platform, which was not limited to pre-selected regions of interest. Similar approaches have been employed to study the DNA methylome of hepatocellular carcinoma (15).
Materials and methods
Electronic database information
The methylation data are available at the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) under series accession number GSE93373.
Blood donors and cell preparations
Blood samples were obtained from 8 unrelated healthy Chinese donors. Mononuclear cells were isolated via density gradient centrifugation using Ficoll-Hypaque. B cells were isolated through positive selection using DynabeadsTM CD19 Pan B magnetic beads (Invitrogen, Thermo Fisher Scientific, MA, USA). An aliquot of B cells was frozen and a second aliquot of B cells was used to establish LCLs by incubating with B95-8 cell supernatant according to standard EBV transformation protocols (16). In brief, B cells were cultured with RPMI 1640 medium containing 20% fetal bovine serum, penicillin and streptomycin and supernatant of the B95-8 cell. The cells were passaged every week until stable growth was exhibited. This study was approved by the Institutional Review Board of the Peking University Cancer Hospital, China. All healthy donors in this study provided written informed consent.
DNA methylation profiling analysis
Genomic DNA was extracted from human primary B cells and LCLs using a DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). The EZ DNA Methylation Gold Kit (Zymo Research, Inc., Irvine, CA, USA) was employed for bisulfite conversion of all DNA samples according to the manufacturer’s protocol. Approximately 600 ng of the bisulfite-converted DNA was analyzed with an Infinium Methylation850K BeadChip (Illumina, San Diego, CA, USA) following the manufacturer’s guidelines at Shanghai Biotechnology Corporation. These chips include more than 850,000 methylation sites across whole genome.
DNA methylation data were analyzed using the methylation analysis module within BeadStudio software employing default parameters (Illumina, Inc., San Diego CA, USA). The probes with low-quality signals (detected P-value >0.05), and those overlapped with single-nucleotide polymorphisms were removed. Finally, probes from X and Y chromosomes were excluded, leaving 707,200 unique probes for analysis. Methylation values, referred to as β-values, were calculated as the ratio of the methylated signal intensity to the sum of the methylated and unmethylated signals after background subtraction. The β-values were reported as a DNA methylation score ranging from 0 (completely unmethylated) to 1 (fully methylated). The overall data quality was examined by calculating mean detection P-values. We considered CpGs to be differentially methylated if they showed a false discovery rate (FDR) of <0.05 and |Δβ|>0.2 between the groups.
Pathway and Gene Ontology (GO) analyses
Pathway enrichment analysis for genes was carried out using the Kyoto Encyclopedia of Genes and Genomes (KEGG) human pathways and modules (www.genome.jp/kegg/download). GO analysis was performed using human GO term associations (www.geneontology.org).
Results
Whole-genome analysis of differentially methylated CpG sites
DNA methylation analysis revealed 87,732 differentially methylated CpG sites between LCLs and primary B cells, representing approximately 12.41% of all sites (Figure 1A). Of the differentially methylated probes, 22.75% showed hypermethylation while 77.25% were hypomethylated (Figure 1B).
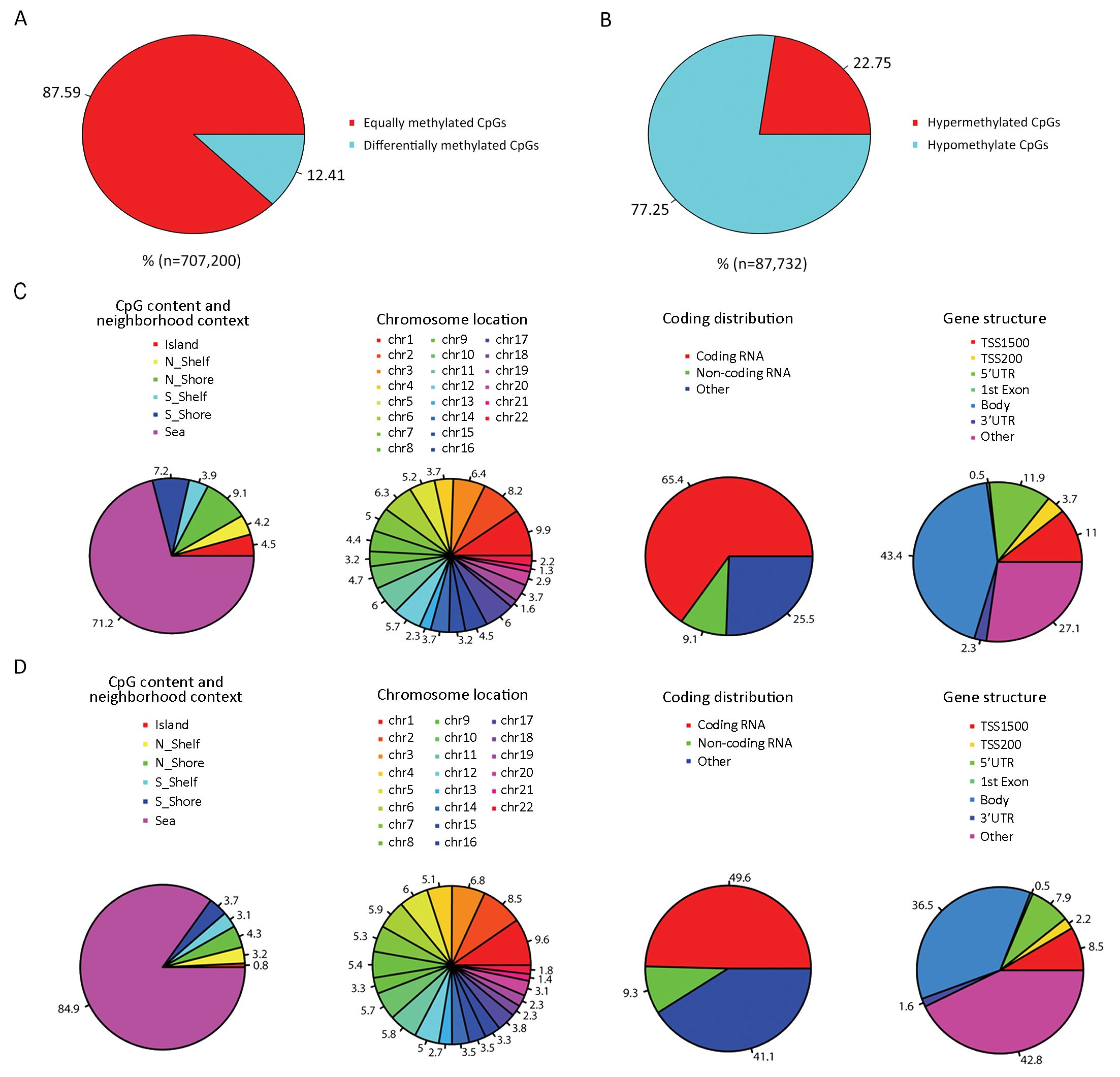
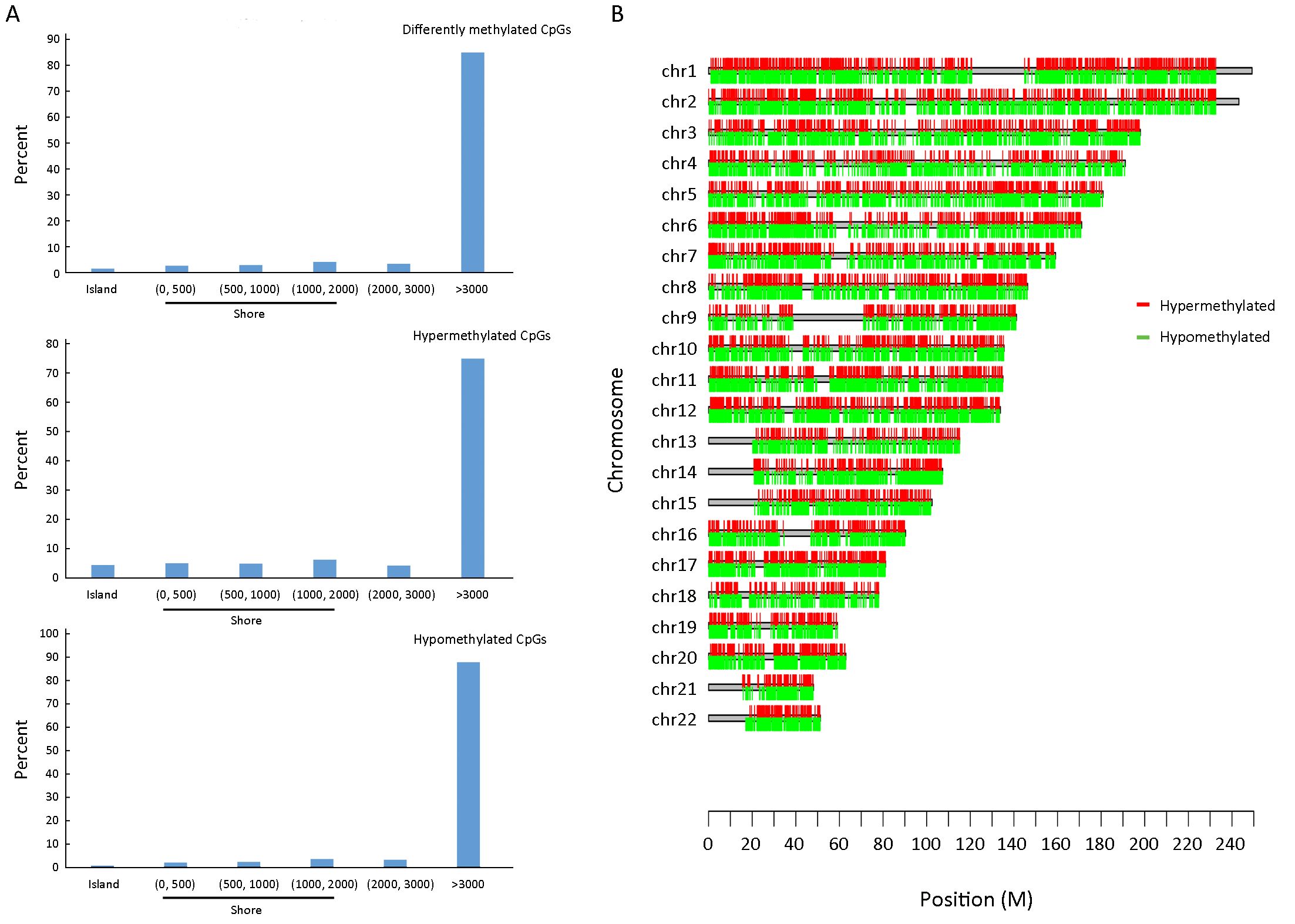
Previous studies have focused on promoters and/or CpG islands, which have been defined as regions with a GC fraction greater than 0.5 and an observed-to-expected ratio of CpG greater than 0.6. However, methylation variations located in not only CpG islands but also CpG island shores both showed strong inverse relationship with differential gene expression. In our study, only 0.8% of hypomethylated sites and 4.5% of hypermethylated sites were located in CpG islands, whereas 8.0% of hypomethylated sites and 16.3% of hypermethylated sites were located in shores (N_shore and S_shore) (Figure 1C, D). In addition, the distribution of differently methylated CpG sites by distance from respective CpG islands showed that DNA methylation variations were relatively evenly distributed in different shore regions (Figure 2A). There was no association between differently methylated CpG sites and chromosome location, and moreover both hypermethylation and hypomethylation variations were relatively evenly distributed in all chromosomes (Figure 1C, D and Figure 2B). Hypermethylated CpG sites were mainly located in coding RNA region (65.4%) and hypomethylated CpG sites were almost half located in coding RNA regions (49.6%). When these differently methylated CpG sites were classified into six categories (TSS1500, TSS200, 5’-UTR, 3’-UTR, body and 1st exon) according to their locations within the genes, no obvious distributional difference was found between hypermethylated and hypomethylated CpG sites (Figure 1C, D).
Discrimination between primary B cells and LCLs by DNA methylation variations
DNA methylation profiles were compared between human primary B cells and corresponding LCLs collected from eight participants. Violin plot showed that mean DNA methylation level was lower in LCLs than in primary B cells (Figure 3A).
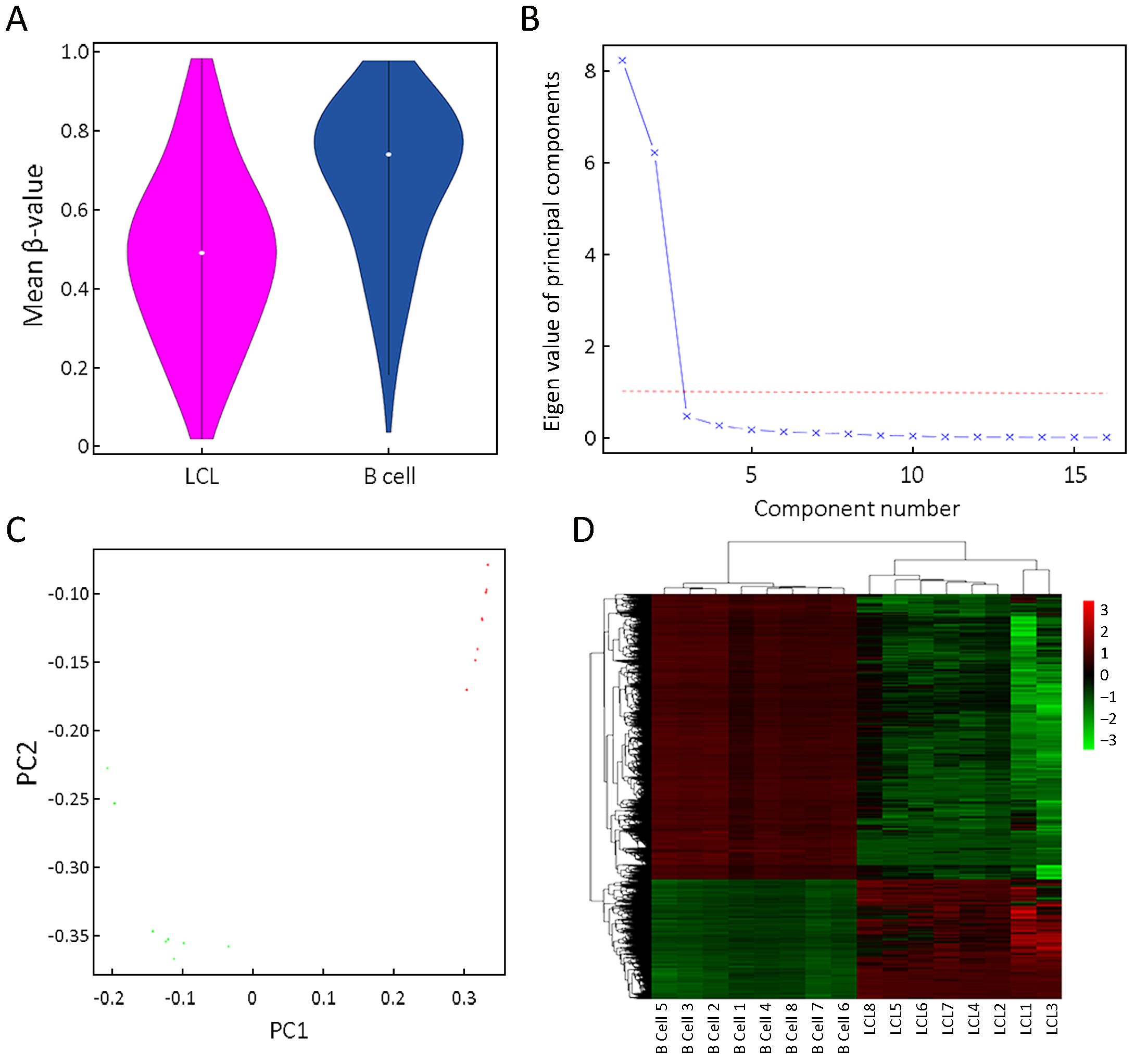
Scree plot showed that the eigen values of the top two principle components (PCs) were more than one, and thus we only included the top two PCs in principle component analysis (Figure 3B). The top two PCs of the DNA methylation profiles, PC1 and PC2, discretely separated all samples into two clusters, primary B cells and LCLs (Figure 3C). In addition, hierarchical clustering analysis also revealed that methylome map could clearly separate primary B cells and LCLs, indicating that EBV-transformed LCLs produced many common methylation variations comparing to primary B cells.
GO and KEGG analysis of differentially methylated genes
To define the functional significance of the extensive changes in the DNA methylation of LCLs, we performed GO analysis to study biological function of the LCL-associated genes with a differently methylated site within 2 kb of their promoter region. The top 20 of significant GO terms for biological processes were shown in Figure 4A. Most of the top GO terms were related to cell activation and immune response, including signaling, cell communication, response to damage, immune response, response to stimulus, inflammatory response, immune system process, defense response, cell activation and cellular response to stimulus.
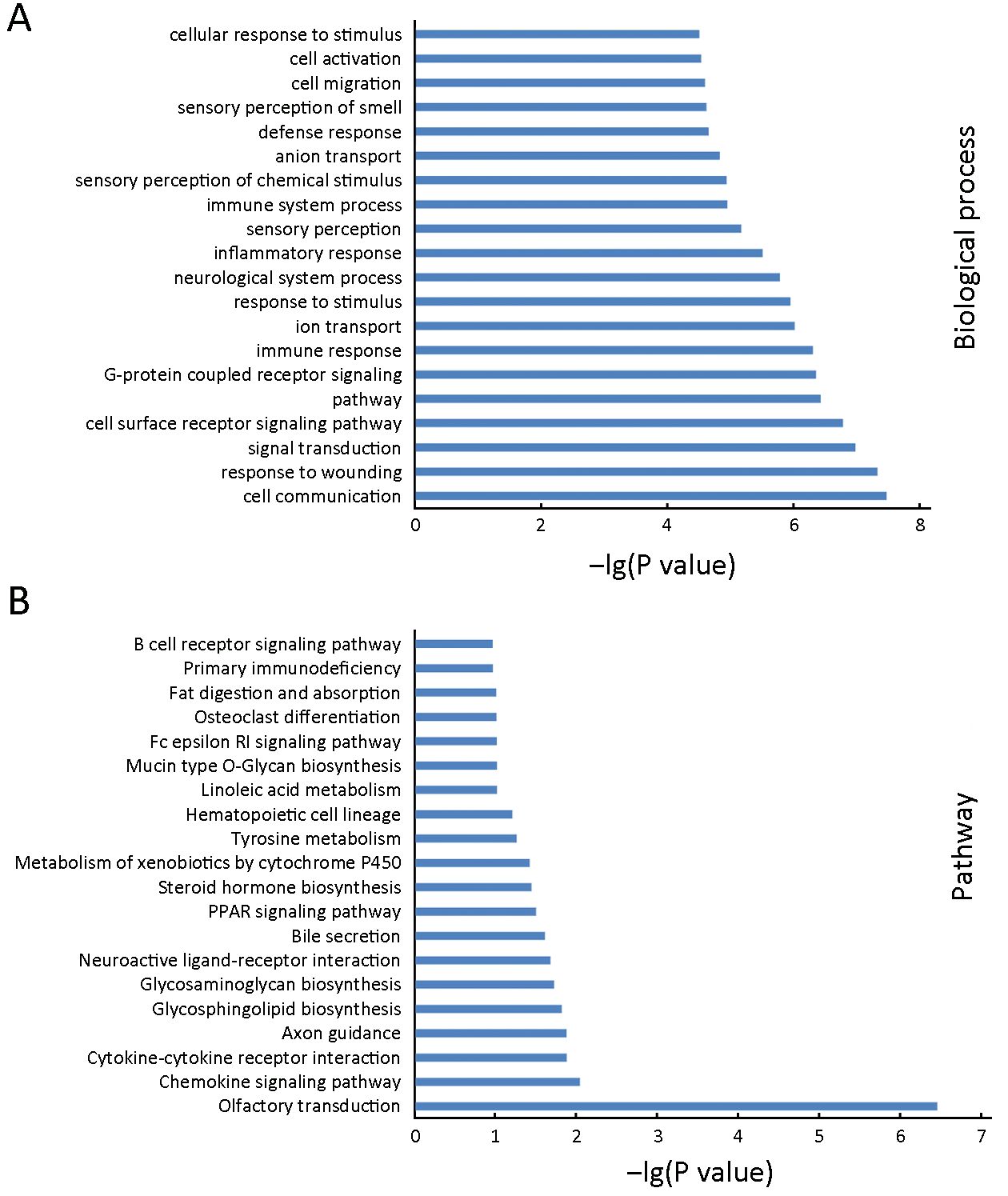
To further investigate key pathways linked to these differentially methylated genes, the significant pathways associated with differentially methylated genes were examined using the KEGG database. Our analysis showed that the differently methylated genes were involved in various cellular pathways, of which chemokine signaling pathway, cytokine-cytokine receptor interaction, peroxisome proliferator-activated receptor (PPAR) signaling pathway and B cell receptor signaling pathway were associated with activation or malignant transformation of human B cells (Figure 4B).
Discussion
Comprehensive comparison of DNA methylome between human primary B cells and matched LCLs is an appropriate model to adequately and comprehensively characterize EBV-induced methylation alterations of B cell, which could give a hint on the methylation mechanism of EBV-induced immortalization. However, most previous studies mainly focused on measuring DNA methylation alterations in promoter or CpG islands, and moreover they did not evaluate biological function and key pathways of differently methylated genes.
In our study, we found that methylation variations occurred in CpG islands were far less than in CpG island shores, and moreover percentage of hypermethylated variations was significantly higher than that of hypomethylated variations both in CpG islands and in CpG island shores. In addition, we found that DNA methylation variations were relatively evenly distributed in different shore regions. Although aberrant methylation variations in CpG island shores besides CpG islands or promoters also play a critical role in many diseases (13,14,17), most previous studies did not describe methylation variations in CpG island shores between human primary B cells and corresponding LCLs in detail (4,8-12). The identification of methylation variations in these regions could open the door to functional studies, such as those investigating the role of differential methylations in CpG island shores.
Functional analysis of these genes associated with differentially methylated sites revealed strong enrichment in the biological process involved in cell activation and immune response, including immune response, immune system process, defense response, cell activation and cellular response to stimulus, which were not reported in previous studies (4,8,9,11,12). This gene enrichment profile indicated that EBV transformation process of human B cells might influence process of cell activation and immune response. In addition, several lines of evidence demonstrated that LCLs showed high levels of expression of the B-cell activation markers and cell-adhesion molecules which were absent or expressed at low levels on resting B cells and yet were transiently induced to high levels when these cells were activated into short-term growth by antigenic stimulation. These findings suggested that EBV-induced immortalization could be elicited through the activation of the same cellular pathways that promoted physiological B-cell proliferation (18,19).
KEGG analysis showed that these differentially methylated genes were involved in various cellular pathways related with activation or malignant transformation of human B cells, such as chemokine signaling pathway, cytokine-cytokine receptor interaction, PPAR signaling pathways and B cell receptor signaling pathway. The cytokine and chemokine families mediate both immune cell recruitment and complex intracellular signaling control mechanisms (20,21), which might play an important role in EBV transformation process of human B cells. However, their effects on EBV-induced immortalization were not systematically evaluated and thus might be an important research area in the future. PPARδ, an important part of PPAR signaling pathway, accounted for many properties of aggressive cancers and was found to enhance Janus kinase (JAK)-mediated phosphorylation of signal transducer and activator of transcription (STAT) proteins in B lymphoma cell lines and primary chronic lymphocytic leukemia (CLL) cells (22,23). In addition, our study also found the enrichment of differently methylated genes in PPAR signaling pathway, which indicated that PPAR signaling pathway could not only promote B-cell cancer cell lines as well as primary CLL cells to survive (22,24), but also be associated with EBV-induced B-cell immortalization. B cell receptor signaling pathway is the key to promote activation and proliferation of B cells and moreover is a driver of lymphoma development and evolution (25,26). Overall, these enrichment pathways of differently methylated genes between primary B cells and matched LCLs indicated that methylation variations of primary B cells could play a major role in EBV-induced B-cell immortalization.
There are some limitations in our study. Our sample size was admittedly limited and thus it would be interesting to enroll more cases. In addition, our study lacked of results about expression of differently methylated genes, which would be evaluated in the future study.
Taken together, our study demonstrated genome-wide DNA methylation variations between primary B cells and corresponding LCLs, which may yield new insight on the methylation mechanism of EBV-induced B-cell immortalization.
Acknowledgements
We thank all of the participants of this study. This work was supported by grants from National Natural Science Foundation of China (No. 81160249, 81301886 and 81760525); Science Foundation of Peking University Cancer Hospital (No. 2017 Zizhu-1); Open Project funded by Key Laboratory of Carcinogenesis and Translational Research, Ministry of Education/Beijing (2017 Open Project-3); Beijing Municipal Science & Technology Commission (No. Z171100001017136); Beijing Natural Science Foundation (No. 7171001); and West China First-Class Discipline Construction Project in Basic Medicine funded by Ningxia Medical University.
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Price AM, Dai J, Bazot Q, et al. Epstein-Barr virus ensures B cell survival by uniquely modulating apoptosis at early and late times after infection. Elife 2017;6:pii:e22509. [PubMed] DOI:10.7554/eLife.22509
- Thorley-Lawson DA. EBV persistence — introducing the virus. Curr Top Microbiol Immunol 2015;390:151–209. [PubMed] DOI:10.1007/978-3-319-22822-8_8
- Skalska L, White RE, Parker GA, et al. Induction of p16INK4a is the major barrier to proliferation when Epstein-Barr virus (EBV) transforms primary B cells into lymphoblastoid cell lines. PLoS Pathog 2013;9:e1003187. [PubMed] DOI:10.1371/journal.ppat.1003187
- Hansen KD, Sabunciyan S, Langmead B, et al. Large-scale hypomethylated blocks associated with Epstein-Barr virus-induced B-cell immortalization. Genome Res 2014;24:177–84. [PubMed] DOI:10.1101/gr.157743.113
- Kalchschmidt JS, Bashford-Rogers R, Paschos K, et al. Epstein-Barr virus nuclear protein EBNA3C directly induces expression of AID and somatic mutations in B cells. J Exp Med 2016;213:921–8. [PubMed] DOI:10.1084/jem.20160120
- Paschos K, Smith P, Anderton E, et al. Epstein-barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog 2009;5:e1000492. [PubMed] DOI:10.1371/journal.ppat.1000492
- Tsai CN, Tsai CL, Tse KP, et al. The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc Natl Acad Sci U S A 2002;99:10084–9. [PubMed] DOI:10.1073/pans.152059399
- Grafodatskaya D, Choufani S, Ferreira JC, et al. EBV transformation and cell culturing destabilizes DNA methylation in human lymphoblastoid cell lines. Genomics 2010;95:73–83. [PubMed] DOI:10.1016/j.ygeno.2009.12.001
- Sun YV, Turner ST, Smith JA, et al. Comparison of the DNA methylation profiles of human peripheral blood cells and transformed B-lymphocytes. Hum Genet 2010;127:651–8. [PubMed] DOI:10.1007/s00439-010-0810-y
- Sugawara H, Iwamoto K, Bundo M, et al. Comprehensive DNA methylation analysis of human peripheral blood leukocytes and lymphoblastoid cell lines. Epigenetics 2011;6:508–15. [PubMed] DOI:10.4161/epi.6.4.14876
- Åberg K, Khachane AN, Rudolf G, et al. Methylome-wide comparison of human genomic DNA extracted from whole blood and from EBV-transformed lymphocyte cell lines. Eur J Hum Genet 2012;20:953–5. [PubMed] DOI:10.1038/ejhg.2012.33
- Caliskan M, Cusanovich DA, Ober C, et al. The effects of EBV transformation on gene expression levels and methylation profiles. Hum Mol Genet 2011;20:1643–52. [PubMed] DOI:10.1093/hmg/ddr041
- Irizarry RA, Ladd-Acosta C, Wen B, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009;41:178–86. [PubMed] DOI:10.1038/ng.298
- Maunakea AK, Nagarajan RP, Bilenky M, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010;466:253–7. [PubMed] DOI:10.1038/nature09165
- Shen J, Wang S, Zhang YJ, et al. Exploring genome-wide DNA methylation profiles altered in hepatocellular carcinoma using Infinium HumanMethylation 450 BeadChips. Epigenetics 2013;8:34–43. [PubMed] DOI:10.4161/epi.23062
- Neitzel H. A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum Genet 1986;73:320–6. [PubMed] DOI:10.1007/BF00279094
- Wu WK, Yu J, Chan MT, et al. Combinatorial epigenetic deregulation by Helicobacter pylori and Epstein-Barr virus infections in gastric tumourigenesis. J Pathol 2016;239:245–9. [PubMed] DOI:10.1002/path.4731
- Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer 2004;4:757–68. [PubMed] DOI:10.1038/nrc1452
- Price AM, Luftig MA. Dynamic Epstein-Barr virus gene expression on the path to B-cell transformation. Adv Virus Res 2014;88:279–313. [PubMed] DOI:10.1016/B978-0-12-800098-4.00006-4
- Turner MD, Nedjai B, Hurst T, et al. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta 2014;1843:2563–82. [PubMed] DOI:10.1016/j.bbamcr.2014.05.014
- Lippitz BE. Cytokine patterns in patients with cancer: a systematic review. Lancet Oncol 2013;14:e218–28. [PubMed] DOI:10.1016/S1470-2045(12)70582-X
- Sun L, Shi Y, Wang G, et al. PPAR-delta modulates membrane cholesterol and cytokine signaling in malignant B cells. Leukemia 2018;32:184–93. [PubMed] DOI:10.1038/leu.2017.162
- Wang D, Fu L, Ning W, et al. Peroxisome proliferator-activated receptor δ promotes colonic inflammation and tumor growth. Proc Natl Acad Sci U S A 2014;111:7084–9. [PubMed] DOI:10.1073/pnas.1324233111
- Zuo X, Xu M, Yu J, et al. Potentiation of colon cancer susceptibility in mice by colonic epithelial PPAR-δ/β overexpression. J Natl Cancer Inst 2014;106:dju052. [PubMed] DOI:10.1093/jnci/dju052
- Havranek O, Xu J, Köhrer S, et al. Tonic B-cell receptor signaling in diffuse large B-cell lymphoma. Blood 2017;130:995–1006. [PubMed] DOI:10.1182/BLOOD-2016-10-747303
- Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010;463:88–92. [PubMed] DOI:10.1038/nature08638