
Tumor immunotherapy: New aspects of natural killer cells
Introduction
Natural killer (NK) cells contribute to the first line of defense against cancer and virus infection as critical components of the innate immune system. This special population (not restricted to a few clones as happened in T or B cells) of lymphocytes as a whole immediately exhibits responsiveness to challenge by a variety of activating signals without prior sensitization. NK cell activation is determined by the balance of signals delivered by activating receptors and inhibitory receptors (1,2). There are many receptors expressed on NK cells that mediate the delivery of these signals. They can be classified by their structure as killer cell immunoglobulin-like receptors (KIRs) and killer cell lectin-like receptors (KLRs) or by their functions as NK cell-activating receptors and NK cell-inhibitory receptors (3-5). Many studies have focused on the structures and functions of these receptors, among which KIRs are currently the best understood and have been shown to predominantly engage with human leukocyte antigen (HLA)-C. KIRs exhibit both a long form, with an immunoreceptor tyrosine-based inhibition motif (ITIM) in the cytoplasmic portion, and a short form, which delivers activating signals by recruiting DAP12 (6,7). Natural killer cell group (NKG) 2 receptors also recognize HLA-I as their ligand. NKG2D is an activating receptor that interacts with major histocompatibility complex (MHC) class I polypeptide-related sequence (MIC) A, MICB and UL16-binding proteins 1-6 (ULPB1-6) (8). NKG2A is an inhibitory receptor that recognizes HLA-E in competition with the activating receptor NKG2C (9). The natural cytotoxicity receptors (NCRs) (NKp46, NKp44 and NKp30) are a group of activating receptors. NKp44 is only expressed on activated NK cells (10). DNAX accessory molecule-1 (DNAM-1) and T cell immunoreceptor with Ig and ITIM domains (TIGIT) are another set of paired receptors that deliver activating signals or inhibitory signals by engaging with the same ligands [polio virus receptor (PVR) or Nectin-C, respectively] (11). FcγRIIIa [also known as cluster of differentiation (CD)16a] is unique among these receptors due to its capacity to bind to the Fc fragment of IgG antibodies, allowing NK cells to mediate antibody-dependent cell-mediated cytotoxicity (ADCC) (12).
Powerful cytotoxicity is another feature of NK cells. Activated NK cells kill targets via multiple approaches, including direct lysis by perforin and granzyme, induction of apoptosis by FasL/Fas or tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)/TRAIL receptors, and the release of cytokines such as interferon (IFN)-γ and TNF-α to activate systemic antitumor immunity indirectly (13-16).
NK cells can also eliminate tumors in indirect manners as “helpers” to promote dendritic cell (DC)-T cell interactions. NK cells activated by two signals (NKG2D and additional inflammatory cytokines such as IFN-α) acquired the helper activity to induce the stable type-1 polarization of DCs (DC1) though the production of IFN-γ and TNF-α. The DC1 induced by such helper NK cells enhance the production of interleukin (IL)-12p70 and intranodal production of C-C motif ligand (CCL) 19, which is essential for anticancer immunity (17,18). It has also been proved that the depletion of NK cells in vivo neutralized the benefit of DC immunotherapy of tumor (19). The cross-talk between NK cells and DCs illustrates the bridging role of NK cells between innate immunity and adaptive immunity (20).
The discovery of memory-like NK cells further extended our understanding of these cells (21). Tissue-specific NK cells show distinct features, indicating that NK cells are a heterogeneous group (22-24). Recently, NK cells were identified as a subpopulation of innate lymphoid cells, and the role of NK cells within the innate immune system has become increasingly well defined (25).
NK cells are believed to be a group of powerful immune effectors in tumor surveillance and control; however, evidence of NK cell malfunctions has been observed in many studies of patients with cancer, and many cancers establish their path to escape immune surveillance through NK cells. Thus, it may be possible to achieve promising clinical outcomes by manipulating NK cells with the intent to reverse their malfunctions, which are due to multiple suppressive mechanisms. Herein, we discuss the current cancer immunotherapies based on NK cell biology. These approaches include adoptive cellular immunotherapy, genetically modified NK cell therapy, therapeutic antibody-derived ADCC, NK cell checkpoint blockade and novel therapies involving bispecific proteins or oncolytic viruses associated with NK cells.
NK cells are critical for tumor immune surveillance
NK cells have been linked to immune responses aimed at the elimination of tumors and viruses since NK cells were first identified. Evidence supporting the hypothesis that malfunctions of NK cells are related to an increased tumor incidence has been found in both clinical researches and animal model studies (26).
NK cells are capable of lysing tumor cells without prior exposure. When challenged by tumors, NK cells are activated in two main ways. Decreased expression of MHC class I on tumor cells activates NK cells through a lack of inhibitory signals, which is known as the “missing-self” theory. Tumor cell stress induces the expression of activating NK cell receptor ligands, which leads to NK cell activation. These activating receptors include NKGD2, NKp30, NKp44, NKp46 and DNAM-1 (11,27-29). Moreover, in several medical scenarios, NK cells can be activated by tumor-associated antigen (TAA)-specific therapeutic antibodies via CD16a-mediated ADCC effects (30). However, this effect in the context of natural tumor immune surveillance is rarely reported (Figure 1).
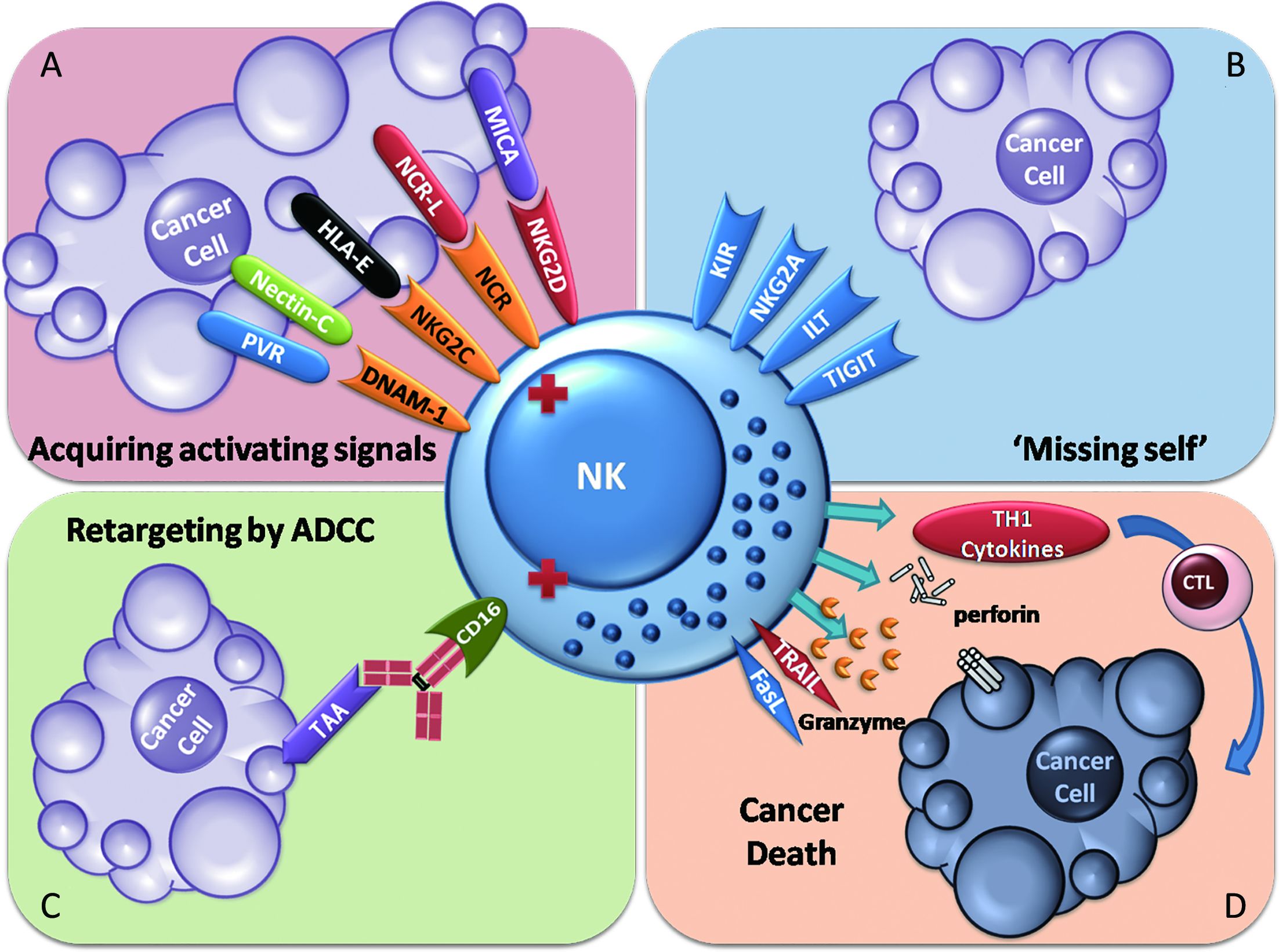
NK cells in healthy individuals are important components of the innate immune surveillance of cancer. Their primary objective appears to be prevention of the metastasis of tumors in the circulation since they are more frequently found in the blood than at tumor sites (31). This phenomenon has additional potential explanations. A high frequency of NK cell accumulation in nascent tumor sites may not be easy to detect, and tissue barriers of solid tumors may prevent NK cells from reaching the tumor core (32,33).
NK cells are functionally inhibited in tumor microenvironment
NK cell deficiency results in an increased incidence of tumors as well as virus infections (34-38). Individuals who exhibit lower NK cell activity are believed to be at higher risk of tumorigenesis. Studies of non-small cell lung cancer (NSCLC) and breast cancer have shown that the function of intratumoral NK cells is impaired compared with peripheral NK cells. Moreover, the developmental stage of NK cells in tumors was found to be more immature compared with peripheral NK cells (39-41). Aging, genetic defects and chronic infection may contribute to lower NK cell activity (32,35,37,42).
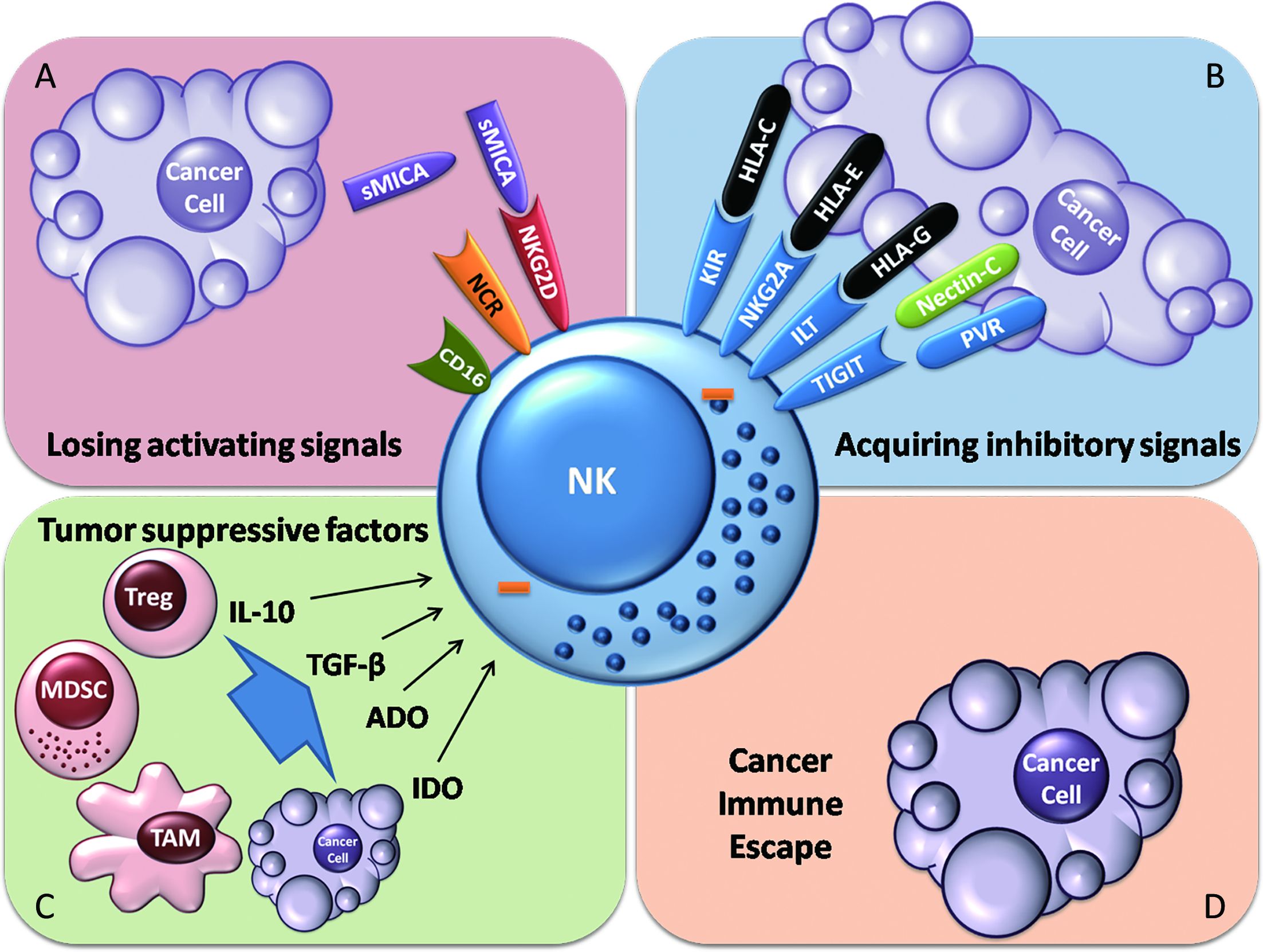
Tumor escape from NK cell immune surveillance predominantly occurs via two mechanisms: reduction of activating signals or increases in inhibitory signals delivered to NK cells. In several studies, soluble NKG2D ligand (NKG2DL) has been observed to be shed from tumor cells, which may prevent NK cells from recognizing tumor cells bearing this ligand (43-45). Chronic stimulation of NKG2D signals may downregulate the expression of NKG2D, which leads to NK cell hyporesponsiveness (46). NK cells exhibit reduced DNAM-1 expression in acute myeloid leukemia, which implies that the expression of NK cell-activating receptors can be downregulated to suppress NK cell activation (47). The expression of ligands of activating NK cell receptors on tumors, such as CD155 (also known as PVR), can be downregulated in tumor environments (48), which is also a protective strategy in which tumor cells preserve the expression of HLA (31).
When tumors escape immune surveillance, tumor evasion occurs. Negative regulatory immune cells and suppressive molecules may help tumors to further suppress NK cells. Myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs) and regulatory T cells (Tregs) are the major types of immune cells that suppress NK cell activation. Ly6Cneg MDCS induced by IL-1β in mice bearing tumors can inhibit NK cell development and antitumor responses by downregulating NKG2D expression on NK cells (49). Surprisingly, cytotoxic T lymphocytes (CTLs) were shown to promote tumor cell-mediated recruitment of MDSCs by Fas signaling (50). TAMs can secrete transforming growth factor (TGF)-β (51). TGF-β inhibits the antitumor activity of NK cells by downregulating the expression of NKG2D, decreasing IFN-γ production and interfering with cytotoxicity (52). Activin-A, which is secreted by dendritic cells (DCs), also inhibits IFN-γ secretion by NK cells but does not interfere with perforin or granzyme B production (53). Additionally, IL-1β secreted by 6-sulfo LacNAc DCs induces NK cell apoptosis (54). Tregs can suppress NK cells through deprivation of IL-2, induction of NK cell death and generation of Indoleamine 2,3-dioxygenase (IDO) and adenosine (ADO) via the expression of CD39 and CD73 (55-57). IDO and ADO may interfere with the signaling of NK cells, which is detrimental to NK cell activation (58-60).
Moreover, several tissue barriers around tumors form a shelter against NK cells. One example of this phenomenon is the fibrotic shield surrounding pancreatic cancer, which prevents immune cells from reaching the tumor core. The existence of such barriers may also explain why NK cells are rarely found at tumor sites (33).
All the above mechanisms employed by tumors suppress NK cell activation (Figure 2). Alterations of the NK cell phenotype can even be observed at tumor sites (39). Therefore, restoring NK cell antitumor activity is critical for establishing host immunity against cancer, which is the primary objective of cancer immunotherapy.
NK cells and cancer immunotherapy
Therapeutic strategy for restoring NK cell antitumor activity
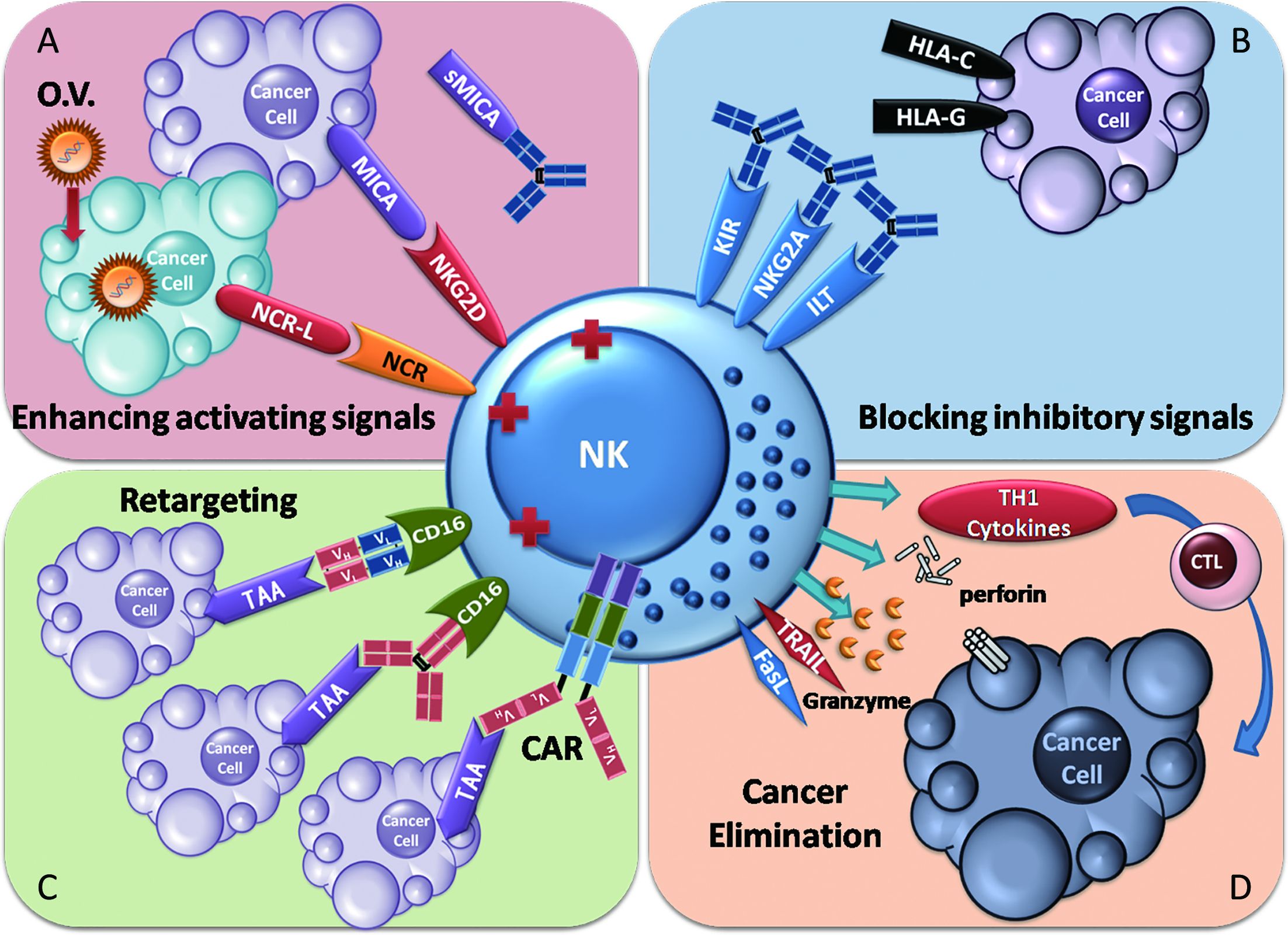
Since NK cell antitumor function is usually impaired in cancer patients, restoring this antitumor function is a primary therapeutic objective. The strategies for restoring NK cell function can be understood as following two parallel paths: increasing factors that activate NK cell functions or decreasing inhibitory factors, according to the fundamental properties of NK cell activation, which is regulated by the orchestrated balance of activating signals and inhibitory signals.
NK cell adoptive cellular immunotherapy provides large amounts of activated NK cells to directly supplement or replace malfunctioning NK cells within cancer patients. Cytokines, such as IL-2, IL-15 and IL-21, are employed to enhance the survival of NK cells ex vivo or in vivo. Retargeting strategies, including the use of therapeutic antibodies, bispecific proteins and chimeric antigen receptor (CAR)-expressing NK cells, have been utilized to enhance the specificity of NK cells for the elimination of cancers. Genetic modification of NK cells and oncolytic virotherapy have also been introduced to facilitate NK cell antitumor activation.
In contrast, blocking NK cell-inhibitory receptors with antibodies minimizes the inhibitory signals delivered to NK cells. Blockade of the soluble ligands of activating receptors may restore the responsiveness of activating NK cell receptors. Haploidentical transplantation reduces inhibitory signals through KIR mismatch.
Thus, NK cell antitumor activity can be restored by multiple approaches. We will review recent studies in these diverse fields (Figure 3).
NK cell-based adoptive cellular immunotherapy
Adoptive cellular transfer therapy with NK cells has been widely studied and practiced, both in experimental models and clinical trials. The fundamental objective of this type of therapy is to revive the patient’s innate immune surveillance and control of tumors via the infusion of large amounts of activated NK cells. NK cell-based adoptive transfer has shown efficacy in treating hematopoietic malignancies. The strategy and clinical trials of NK cell-adoptive transfer have been specifically reviewed elsewhere (61). Herein, we will briefly review the current strategy of NK cell-based adoptive cellular immunotherapy and discuss some of the remaining questions in this field.
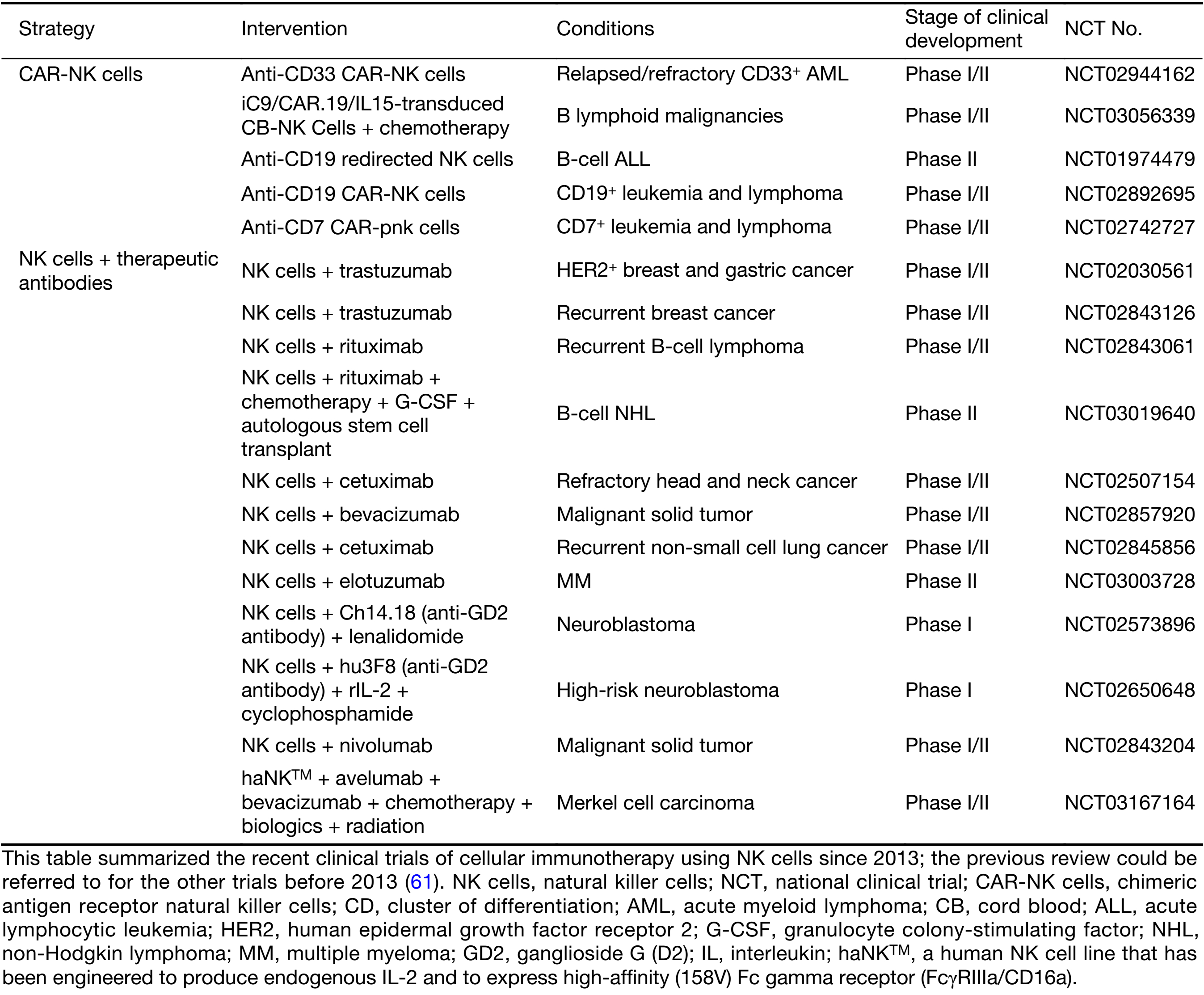
There are four primary sources of NK cells for adoptive transfer: autologous NK cells, allogeneic NK cells, NK cell lines and genetically modified NK cells. The use of autologous NK cells is the most primitive strategy for NK cell adoptive transfer and involves the clinical practice of lymphokine-activated killer cell (LAK) induction by IL-2 in vivo. Due to safety concerns regarding the toxicity induced by the systemic administration of IL-2, in addition to the limited antitumor efficacy achieved, this approach has been abandoned. As an alternative to this strategy, ex vivo-expanded autologous NK cells have been used. Allogeneic NK cells are another source for NK cell adoptive transfer. Among the approaches of allogeneic NK cell adoptive transfer, haploidentical transplantation has attracted attention due to its efficacy in treating hematopoietic malignancy due to the KIR mismatch, which increases the susceptibility of tumors to NK cell-mediated killing (62). NK cell lines are another option as a source of NK cells. The advantages of NK cell lines in adoptive transfer are their stability and the quantity of therapeutic cells obtained. As cell lines, they share similar qualities, which facilitates the quality control of Good Manufacturing Practice (GMP) products. The proliferation capacity of these cell lines in vitro provides the quantity of cells required for clinical demands. There are a number of established NK cell lines, among which NK-92 is the most promising and has been applied in clinical trials with the approval of the U.S. Food and Drug Administration (FDA) (63). Recently (in 2017), activated NK therapy was qualified as an orphan drug treatment for Merkel cell carcinoma by the FDA (reported by NantKwest Inc.). NKG was a newly established cell line in China, with promising antitumor responsiveness (64). Genetically modified NK cells are the newest generation of NK cell-adoptive transfer therapy. By manipulating NK cells genetically, one can create more efficient antitumor NK cells with autocrine cytokines, over-expression of activating receptors, reduction of inhibitory receptors and NK cells armed with targeting components, such as CARs. CAR-NK is reviewed in the next section.
Except for NK cell lines, the expansion of NK cells ex vivo is a fundamental step for clinical practice. There are many sources for expanding NK cells ex vivo, and the most favorable is peripheral blood mononuclear cells (PBMCs). The final expansion rate and purity of NK cells are the key parameters of NK cell expansion using PBMCs. Enriching NK cells via isolation before culturing for expansion is an optional step that may increase purity but limit the expansion rate; however, expansion without prior isolation may increase the expansion rate but lead to relatively lower purity (65). There are also approaches for acquiring NK cells from umbilical cord blood (UCB) and induced pluripotent stem cells (iPSCs), which were reviewed previously (61).
The stimulators are the other critical factors to activate and expand NK cells ex vivo. Combinations of cytokines or feeder cells have been used to expand UCB-derived NK cells (66-69). Feeder cells engineered with membrane-bound IL-15 and 4-1BB significantly increase the expansion of NK cells with high purity (70). Anti-CD16 or anti-CD52 antibodies also strongly expand NK cells (71,72).
Given the promising features of immune cell adoptive transfer, these cells are referred to as live drugs. The clinical application of this “drug” must therefore meet the basic rules for drugs in terms of safety, efficacy and quality control. Regarding safety concerns, the standard method to assess of remaining feeder cells in therapeutic cellular products should be established. More preclinical and clinical studies on the tumorigenicity of NK cell lines are required, although it has been shown that NK-92 is quite safe in preclinical studies and phase I clinical trials (73). The safety issues examined in CAR-T cell clinical trials should also be assessed for CAR-NK. For efficacy assessment, sensitive approaches should be developed to assess the antitumor activities of NK cells and their duration. Statistically significant efficacy of NK cell therapy in large populations has yet to be observed. As these cells are designed for clinical application, equivalent efficacy should be considered as one feature of quality control, meaning that every patient should receive therapeutic NK cells showing similar antitumor activity before administration, regardless of individual conditions that might impact the outcome. The application of NK cell lines is quite successful in this regard.
Although promising results for NK cell adoptive transfer have been reported in preclinical and clinical studies, there is still a long way to go to transition between the bench and the bedside.
Genetic modification of NK cells
Genetic modification is a promising approach for arming NK cells with multiple components, with the intent to enhance NK cell antitumor activity genetically. The current strategies for such genetic modification include cytokine gene transfer (e.g., IL-2, IL-12, IL-15 and stem cell factor) to promote NK cell survival, in earlier studies, as well as CAR arming for retargeting TAA expressed on tumor cells and high affinity CD16a arming for improving ADCC effects in more recent studies (61,74-77).
CAR-NK is a type of CAR-gene-modified NK cells to redirect NK cell to kill tumor, with a similar goal to CAR-T. For T cells with specific immunity, natural specificity for tumors depends on a complicated process of antigen presentation and recognition, which is vulnerable to tumoral immune escape. Arming T cell with a CAR simplifies the generation of the specificity of their antitumor responsiveness into only one step. Since tumoral heterogeneity is a well-known cause of immune escape (78-80), the specificity to only TAA-bearing tumors of CAR-T cells creates a new limitation for its antitumor responsiveness. However, CAR-NKs acquire a broader antitumor spectrum under a similar scenario. CARs facilitate the ability to focus NK cells on eliminating the major population of tumor cells as tumors bearing no CAR-specific TAAs may be recognized by NK cells with the natural NK cell-activating receptors (74). Thus, CAR-NK might confer a lower risk of immune escape. The major sources of CAR-NKs are NK-92 cell lines and primary NK cells. NK-92 is a promising candidate for application under the CAR-NK strategy, due to its potential to be an “off-the-shelf therapeutic” (63). Many ongoing studies are investigating CAR retargeting for various TAAs, such as CD19, CD20, GD2, human epidermal growth factor receptor 2 (HER2), epidermal growth factor receptor (EGFR), epithelial cell adhesion molecule (EpCAM) and CD138 (81-87). Furthermore, there are additional cell sources for constructing CAR-NKs. Studies on CAR-NK retargeting for carcinoembryonic antigen (CEA) and CD33 are based on the NK cell line YT (88,89). Another clinical trial of CD33-specific CAR-NKs based on NK92 cells is ongoing (Table 1). These CAR-NKs have shown promising efficacy in the control of tumors in preclinical studies; however, clinical efficacy remains to be seen. Regarding safety concerns about NK-92, based on the cell line background involving EBV infection, it is necessary to irradiate these cells before adoptive transfer (31). Regrettably, loss of CD16 prevents the combination of this technique with therapeutic antibodies. CAR-NKs based on primary NK cells have also been reported (90). This CAR-NK strategy may preserve the natural capacity of the cells to respond to therapeutic antibodies; however, quality control of this population remains to be studied.
Full table
Another approach for genetic modification is to arm NK cells with a high-affinity variant of CD16a. Details of this variant of CD16a are described in the following section. Distinguished from NK-92 cells which lack CD16a expression (63), NK-92Fc was generated with high-affinity CD16a and has shown promising efficacy in ADCC. However, CAR-NK is functionally superior to NK-92Fc in combination with therapeutic antibodies that are specific to the same TAA in vitro and in vivo (75).
Reducing the expression of inhibitory NK cell receptors via genetic modification is another possible strategy to enhance NK cell antitumor function. NK-92 cells may have such potential, although they are not typically genetically modified. NK-92 cells transfected with gene-encoded KIRs show lower cytotoxicity. However, the possibility of hyporesponsiveness due to deletion of inhibitory NK cell receptors always exists.
Nevertheless, the most important limitation of the genetic modification of NK cells is their efficient transduction. The application of retroviral vectors facilitates this approach, such as the optimized transduction with alpha-retroviral vectors (91).
In summary, genetic modification provides more options for engineering NK cells with infinite possibilities, albeit with important considerations.
Combination immunotherapy and NK cells
ADCC mediated by NK cells and tumor-targeted therapeutic antibodies
Over the last few decades, therapeutic antibodies have come to represent a typical class of biologics exhibiting an impressive ability to suppress cancer, and many of these antibodies have already been approved by the FDA (92,93). Many different types of antibodies have been applied in clinical treatments with favorable outcomes in many cases (94). These antibody therapies can kill tumor cells directly by blocking growth factor signaling and inducing apoptosis, by suppressing angiopoiesis and stromal cells, or by inducing antitumor immunity (92). There are three major mechanisms through which therapeutic antibodies enhance immune-mediated tumor cell killing: TAA targeting, immune inhibitor blockade and immune effector stimulation. TAA-targeting therapeutic antibodies bind TAA via the Fab fragment and induce an immune response via the Fc fragment. The therapeutic efficacy of these antibodies depends on immune effectors, such as ADCC (mainly through NK cells), complement-dependent cytotoxicity (CDC), and opsonization by phagocytes, according to in vitro studies. Immune inhibitors that cause cancer immune escape have recently become a major focus of cancer immunotherapy, including immune checkpoint proteins, such as programmed cell death protein 1 (PD-1), and suppressive agents in the tumor microenvironment, such as adenosine (ADO) (95,96). Blocking the suppressive signaling by these inhibitors partially rescues the viability of immune surveillance and provides clinical benefits.
The precise mechanisms underlying the therapeutic effects of many of the TAA-targeting antibodies are unclear. However, preclinical research has shown that some of them are critically dependent on the ADCC effect (30). ADCC is mediated by Fc receptors (FcγRIIIa). NK cells have been suggested to be the key effectors of ADCC, since only NK cells express the activating Fc receptor CD16a without expressing FcγRIIb, which transmits inhibitory signals to other Fc receptor-bearing immune cells (30). Among the available targeting therapeutic antibodies, rituximab and trastuzumab are efficacious and target CD20 and HER2, respectively (46,97,98). It has been shown that a mutant Fc fragment antibody targeting HER2 loses the capacity to suppress cancer in vivo (99). In human studies, the therapeutic efficacy of trastuzumab in treating metastatic breast cancer has been demonstrated to be correlated with NK cell activity; however, the treatment duration is controversial (100,101). FcγRI/III deficiency in mice impairs the therapeutic efficacy of an anti-GD2 antibody (102). Many studies have focused on the ADCC effect of targeting antibodies, such as rituximab, obinutuzumab, trastuzumab, cetuximab and elotuzumab (103-112) (Table 2). Notably, the action of elotuzumab in the treatment of multiple myeloma is two-fold: it induces ADCC by binding to SLAMF7 expressed on multiple myeloma cells, and it activates NK cells by binding to SLAMF7 expressed on NK cells (113).
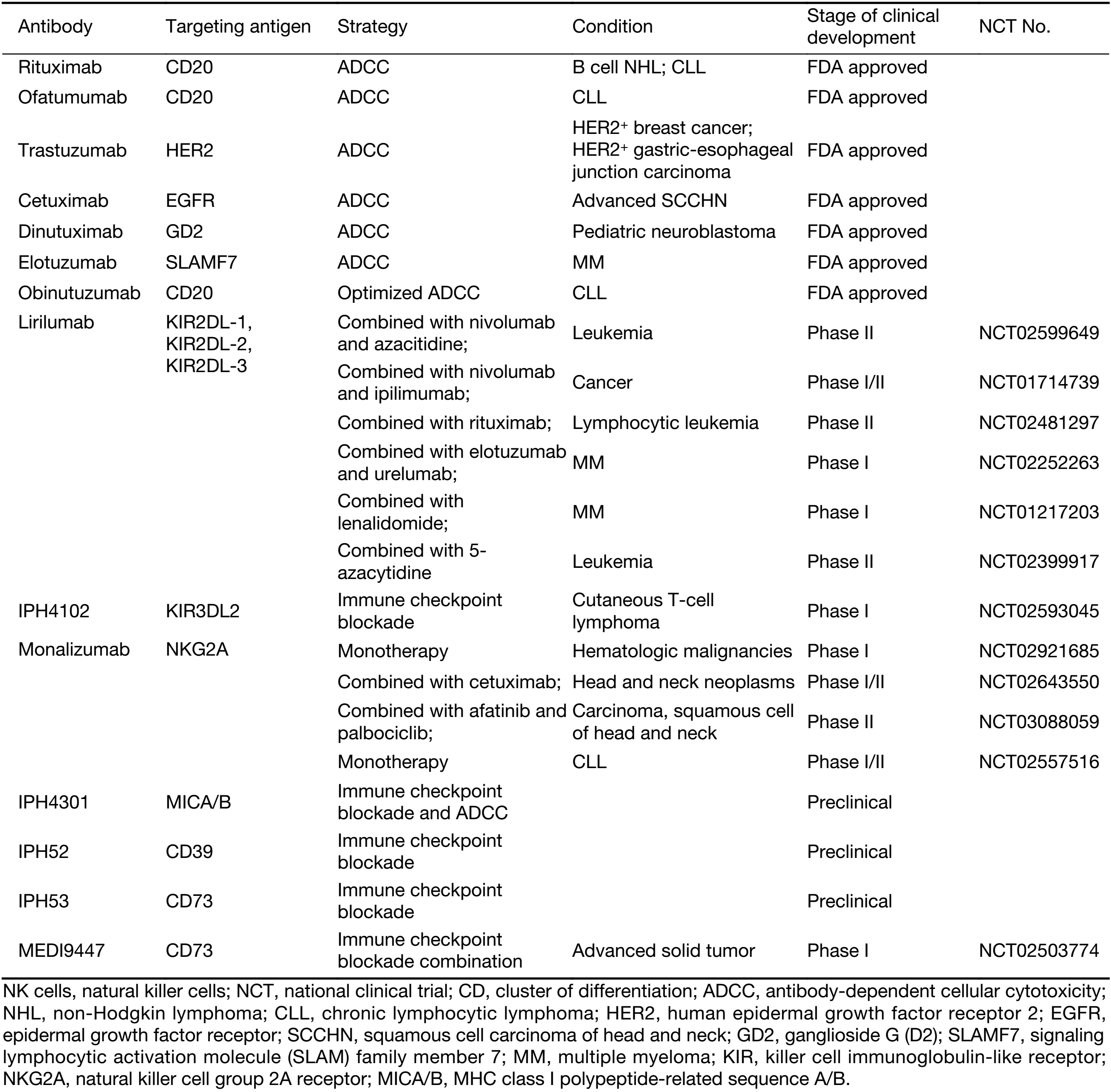
Full table
The discovery of polymorphism in FcγRIIIa further demonstrated the promising role of NK cells in ADCC. FcγRIIIa-158V is an allotype that results from a single nucleotide polymorphism at amino acid 158, resulting in a higher affinity for IgG antibodies. The lysis of CD20+ tumor cells induced by rituximab treatment is much higher in FcγRIIIa-158V/V donors than in FcγRIIIa-158F/F donors, who carry another allotype of FcγRIIIa with a lower affinity for IgG antibodies (114).
Additional NK cell receptors (either activating or inhibitory) are reported to impact NK cell activation mediated by CD16a. Activating receptors involving NKG2D signaling enhance the efficiency of ADCC, while HLA-mediated signaling may limit the efficacy of therapeutic antibodies (115-120). However, modification of the Fc fragment may decrease the inhibitory effect via the KIR/HLA interaction, and one study indicated that KIR inhibitory signaling can be overcome by ADCC mediated by therapeutic antibodies (120,121).
As NK cells are key effectors in the process of ADCC, adoptive transfer with ex vivo-expanded and activated NK cells combined with therapeutic antibodies improves the ADCC effect in patients with poor NK cells compromised by tumor conditions. In several clinical trials, TAA-targeting antibodies were administered in combination with NK cell infusion (Table 1).
Another strategy for enhancing NK cell-mediated ADCC is to engineer antibodies to optimize their binding affinity to FcγRIIIa, which are known as third-generation monoclonal antibodies (30). Obinutuzumab is a notable example of this type of antibody (104).
One phenomenon observed in therapeutic antibody-induced ADCC should be noted. Continual engagement of NK cell receptors by their ligands results in downregulation of activated NK cell receptors (46). Chronic administration of an anti-CD20 antibody was found to impair NK cell-mediated cytotoxicity via CD16 (122). This chronic stimulation by therapeutic antibodies induces NK cell hyporesponsiveness, which is assumed to be one of the reasons for the development of clinical resistance (46).
Still, a precise understanding of NK cell-mediated ADCC for targeted antibody therapy remains to be obtained.
Restoration of NK cell activity by immune checkpoint blockade
Blockade of immune checkpoints is one promising therapeutic strategy for cancer, and impressive clinical outcomes have been associated with anti-CTLA-4 and anti-PD-1 administration. Since the antitumor activity of NK cells can also be reduced by signaling from inhibitory receptors, inhibitory NK cell receptor antagonists have been developed to enhance NK cell antitumor activity. These studies focus on the major inhibitory receptors of NK cells, including KIR, NKG2A and ILT (Table 2).
Anti-KIR: KIRs inhibit NK cell activation by engaging HLA. Therefore, KIR antagonism is one promising strategy for blocking NK cell checkpoints. It has been shown that the variability in KIR genes predicts the response to anti-EGFR monoclonal antibodies (123). The fully human monoclonal anti-KIR antibody lirilumab (also known as 1-7F9, IPH2101 or IPH2102) was generated to block the three major KIRs (KIR2DL-1, -2 and -3) (124) and was shown to boost the cytotoxicity mediated by NK cells in HLA-matched acute myeloid lymphoma (AML) blasts, both in vitro and in vivo (125). In combination with anti-CD20, lirilumab treatment enhanced NK cell antitumor activity in vitro and in vivo (126). As reported based on two phase I clinical trials, lirilumab is well-tolerated in the treatment of AML and MM (125,127). However, a phase II clinical trial of lirilumab monotherapy for the treatment of elderly patients with AML did not meet the primary efficacy endpoint (according to an announcement by Innate Pharma). This result is not encouraging, implying that NK cell checkpoint blockade therapy is much more complicated than expected and that therapeutic efficacy will depend on more than a single agent that blocks only a few types of inhibitory receptors. Combined therapy studies with lirilumab are ongoing with multiple agents (Table 2). One clinical trial of lirilumab combined with lenalidomide for the treatment of relapsed/refractory MM recorded five serious adverse events (128). The preliminary efficacy of another clinical trial of lirilumab in combination with nivolumab for the treatment of squamous cell carcinoma of the head and neck showed that the objective response rate was 24%, without additional toxicity (according to reports from Innate Pharma). It has also been found that the effect of daratumumab-mediated ADCC in the killing of MM cells is enhanced by lirilumab ex vivo (129), which suggests that blockade of KIR may help to maintain the required ADCC effect, which is critical for the efficacy of many therapeutic antibodies. Moreover, another KIR-specific monoclonal antibody, IPH4102, which is specific to KIR3DL2, has been generated. KIR blockade therapy combined with KIR-mismatched NK cell transplantation and TAA-targeting therapeutic antibodies provides a potential solution to improve NK cell responsiveness. This strategy has been verified in an experimental model of mixed-lineage leukemia 1 (MLL)-rearranged leukemia, which is NK cell resistant (121). This study indicated that the variability of KIR expression in individuals is a limiting factor for efficacy of KIR blockade therapy, and activating factors play a critical role as well.
Anti-NKG2A: Another important inhibitory receptor of NK cells is NKG2A, which forms a disulfide-linked heterodimer with CD94. NKG2A belongs to the C-type lectin family, along with NKG2C and NKG2D. NKG2A recognizes HLA-E in competition with NKG2C, which is an activating receptor of NK cells. NKG2A was shown to inhibit NK cell antitumor function in multiple tumor types (9,130). Therefore, NKG2A is a potential candidate for checkpoint blockade therapy. The NKG2A-blocking antibody named monalizumab (also known as IPH2201) has been generated. Several clinical trials of monalizumab are ongoing (Table 2). As a monotherapy, monalizumab exhibits no dose-limiting toxicity (as reported by Innate Pharma). Further evaluation of its efficacy is ongoing. The strategy of combination therapy with monalizumab is similar to that for lirilumab.
ILT: The immunoglobin-like transcript (ILT)/leukocyte immunoglobin-like receptor (LIR) is another NK cell-inhibitory receptor, which recognizes HLA-G as its ligand. HLA-G is expressed on fetal cells to maintain maternal-fetal tolerance of NK cells; however, it is also detectable in a variety of cancers (131). Blockade of LIR-1 did not enhance NK cell-mediated cytotoxicity toward multiple myeloma cells (132), which is a reflection of the lack of knowledge about the influence of ILT in tumor pathogenesis. However, ILT might still be a potential therapeutic target of immune checkpoints, and the inhibitor of ILT could be developed as a component of combination therapy in the future.
Interestingly, one study found that dual blockade of NKG2A and LIR-1 increases the cytotoxicity of KIR-negative NK cells (133). This report implied that inhibitory NK cell receptors may function in different subsets of NK cells or in distinct states of differentiation. Human NK cell differentiation can be phenotypically characterized as follows: CD56brightCD16dimCD57–KIR–NKG2A++, CD56dimCD16dimCD57–KIR–NKG2A++, CD56dimCD16brightCD57–/+KIR+NKG2A+, and CD56dimCD16brightCD57+KIR+NKG2A– (134). During differentiation, the inhibitory receptors of NK cells change dynamically, with acquisition of KIR and loss of NKG2A being observed. Thus, the heterogeneity of NK cells should be considered in therapeutic strategies. Nearly half of the NK cells in circulation are KIR negative (133), and treatment with lirilumab will fail if this KIR-negative NK cell population is responsible for tumor elimination when tumor cells express high levels of HLA-E. More precise profiling of the type of cancer based on biomarkers and the phenotypes of NK cells in patients should be performed whenever NK cell-based immunotherapy is conducted.
Anti-TIGIT: TIGIT is a particularly distinctive inhibitory NK cell receptor that is also expressed on CD8+ T cells and NKT cells, since the TIGIT ligand does not belong to MHC class I, in contrast to the other three inhibitory NK cell receptors mentioned above. Instead, it typically recognizes PVR, Nectin-2 or Nectin-3 as a ligand (11). These ligands are over-expressed on cancer cells. However, most of the available evidence indicates that TIGIT is a characteristic of “exhausted” T cells in cancer and is functionally linked to NK cells in auto-immune diseases and inflammation control (11,95,135). Thus, the therapeutic application of a TIGIT-blocking antibody is focused on reviving T cell responsiveness, rather than NK cells at present.
Anti-sMICA: This type of blockade therapy exhibits a distinguishing characteristic. The shedding of soluble MICA/B from tumor cells is a well-known mechanism of immune escape from NK cells (8,136). It then blocks NKG2D activity, leading to downregulation of NKG2D expression (8,31). Thus, a MICA/B-blocking antibody (IPH4301) provides a path to overcome the suppression of NKG2D signaling by soluble NKG2D ligands. This strategy theoretically involves two mechanisms: blocking soluble MICA/B and inducing ADCC effects on tumor cells expressing MICA/B.
Anti-CD39 and anti-CD73: IDO and ADO are well-known inhibitors of NK cell activity (137). IDO can suppress the expression of NKG2D and NKp46 on NK cells and reduce the cytotoxicity of NK cells (60). ADO has also been found to impair NK cell antitumor activity (138). CD39 and CD73 are membrane-bound extracellular enzymes that catalyze the transformation of ATP to AMP, which is upstream of IDO and ADO. They are expressed on regulatory immune cells (e.g., Tregs) and several types of tumor cells (57,139). CD73 expression on tumor-infiltrating NK cells is associated with suppression of NK cell activity (57). CD39- and CD73-blocking antibodies have been generated, and both anti-CD39 and anti-CD73 increased the cytotoxicity of NK cells toward ovarian cancer cells in vitro (140). These CD39- and CD73-blocking antibodies represent another pair of promising candidates for immune checkpoint blockade therapy to restore NK cell activity.
Under the scenario of T cell activation, inhibitory signals such as CTLA-4 always arrive after activating signals in a sequential manner. Blockade of T cell-inhibitory receptors is sufficient to restore the already existing activation. Distinct from adaptive immunity, activation of NK cells depends on the balance of signals delivered by activating receptors and inhibitory receptors. Blockade of inhibitory signals alone will not rescue NK cell activity to kill cancer, which will only succeed if combined activating signals occur simultaneously. These findings suggest that the strategy of NK cell immune checkpoint blockade may fully function as part of combination immunotherapies.
Since NK cell responsiveness is fine-tuned by stimulation with appropriate inhibition throughout an individual’s lifetime, according to the license theory, prolonged blockade of NK cell-inhibitory receptors may induce hyporesponsiveness of NK cells (2,46). A more precise investigation should be performed to illustrate the pharmacological dynamics of blocking antibodies to avoid NK cell hyporesponsiveness.
Using agonist antibodies for augmentation of NK cell antitumor activity
IL-2, IL-12, IL-15, IL-18 and IL-21 are well-established cytokines that are critical for NK cell differentiation, proliferation or survival (141-146). IL-2 was once considered as a promising therapeutic biologic for cancer treatment, as it was shown to trigger LAKs to kill cancer in a clinical setting (147). However, it fell out of favor due to its potential to induce Treg proliferation (148,149). The other cytokines are still in preclinical studies, and there are concerns about their safety. Hence, clinical application of these cytokines should proceed cautiously. As these cytokines exhibit multiple functions in the immune system, it is rarely possible to harness them with only an isolated impact on NK cells and no unwanted systemic adverse effects, including potentially severe toxicity.
Interestingly, several pharmacological agents (such as lenalidomide, histone deacetylase, demethylating agents, DNA-damaging agents, bortezomib, histamine, imatinib and sorafenib) are capable of restoring NK cell activity (52). Some of these agents can induce the activation of NK cell receptor ligand (histone deacetylase, demethylating agents, DNA-damaging agents) or Fas and TRAIL receptor (bortezomib) expression on cancer cells. Some of them can induce activating NK receptor expression on NK cells (lenalidomide). And some of them improve NK/DC cross-talk to activate NK cells (imatinib). Among these agents, lenalidomide has already been used in the clinic, showing indirect and unclear effects on NK cell activation; histamine prevents the downregulation of NKp46 and NKG2D by phagocyte ROS production (52). One super agonist of IL-15 (known as ALT-803) could enhance NK cell cytotoxicity against tumors (150). The dimethyl fumarate metabolite monomethyl fumarate augmented the cytotoxicity of CD56+ NK cells against tumor cells (151).
In contrast to the blocking antibodies of inhibitory NK cell receptors, there are several potential agonist antibodies that are applicable for the restoration of NK cell antitumor activity. The 4-1BB (also known as CD137) agonist antibody enhances tumor-specific cytotoxicity, and the co-membrane-bound forms of IL-15 and 4-1BBL on K562 cell lines significantly induce NK cell expansion (70,152,153). This finding suggests a promising role of 4-1BB in NK cell activation. CD16a and NKG2D are strong activating receptors on NK cells with the potential to overcome inhibitory signals from HLA class 1 members (121,154,155). Therefore, they are target candidates of NK cell agonists. Since CD16a and NKG2D are not expressed on NK cells alone, application of pan-stimulants of CD16a and NKG2D may cause unknown severe systemic toxicity. One example of severe toxicity induced by pan-stimulants is provided by anti-CD28, as multiple organ failure occurred in all six healthy subjects in one clinical trial aimed at evaluating the safety of anti-CD28 in 2006 (156). Preclinical research had demonstrated the safety of this super agonist. However, it was not considered that humans commonly exhibit high levels of memory lymphocytes, which are hyper-responsive to this super agonist. Thus, one can never be too cautious when assessing the risk of pan-stimulants. Nevertheless, there is another option for employing CD16a and NKG2D agonists. Engineered bispecific antibodies and fusion proteins, which are reviewed in the next section, can decrease this risk.
Using bispecific proteins as engagers for activating NK cell receptors and tumor antigens
One feature of the adaptive immune system that distinguishes it from the innate immune system in eliminating tumor cells is the specific recognition of a tumor target. This characteristic creates a more effective elimination pattern for specific pathogens. Thus, it is reasonable to generate NK cells with specific targeting potential to arrest tumor cells more effectively. One of the strategies for meeting this objective is to generate NK cell-associated bispecific antibodies (bsAbs).
Recently, many bispecific antibodies targeting NK cells have been produced. Like all the other bsAbs, these NK cell-associated bsAbs display 2 binding specificities. One binding fragment specifically links to an NK cell-activating receptor, and the other specifically links to a TAA. They are also known as bispecific killer cell engagers (BiKEs), which promotes NK cell-mediated killing. Theoretically, all NK cell-activating receptors could serve as targets. However, only CD16A has been selected thus far, probably due to the successful construction of the anti-CD16A antibody 3G8, which is a functional antibody agonist. CD16A is an activating receptor with low affinity for the Fc fragment of immunoglobulin. It is responsible for ADCC mediated by NK cells. This receptor connects innate immunity with adaptive immunity via the engagement of IgG and incorporates the specificity of NK cells. The fragments showing specificity against TAAs include CD30 [Hodgkin lymphoma (HL)], CD33 [myelodysplastic syndromes (MDS)], CD133 (colorectal cancer), EpCAM (carcinoma), HER2 (breast cancer), EGFR (EGFR-expressing cancer) and CEA (colon carcinoma) (157-164). These bsAbs have shown efficacy in promoting NK cell antitumor activities. CD16xCD33 includes a novel double target with a single specific binding pattern. This bsAb induces NK cell antitumor functions by linking them to both CD33+ MDS target cells and CD33+ MDSCs. CD33+ MDSCs have been shown to act as negative modulators of NK cell antitumor functions. Thus, CD16xCD33 induces NK cells to engage with tumor targets and to disrupt suppression by MDSCs (160).
Another group has extended NK cell-associated bsAbs to trispecific and tetraspecific antibodies. The original version was CD16xEpCAM, which targets human carcinomas (161). Considering that CD16xEpCAM only induces NK cell antitumor cytotoxicity, and not NK cell expansion, an IL-15 cross-linker was incorporated into the bsAb to improve the activation, proliferation and survival of NK cells (165). Furthermore, this trispecific antibody was improved to a tetraspecific antibody, with additional specificity against CD133, which is believed to be a marker of cancer stem cells (166).
CD30xCD16A, also known as AFM13, is a tetravalent bsAb (167). This antibody has already been applied in clinical trials (NCT01221571, NCT02321592 and NCT02665650) to treat HL. CD30xCD16A showed good safety and tolerance in a phase I trial and displayed an overall disease control rate of 61.5% (168). Importantly, the impaired functions of NK cells from HL patients were shown to be rescued by CD30xCD16A, which was explained by the impaired NK cell function resulting from the soluble NKG2D ligand or NKp30 ligand. CD16A provides an activation pathway that is independent of NKG2D and NKp30 (157). However, data from NSCLC patients indicate that CD16 expression on NK cells may also be downregulated in certain cancer types or cases, and CD16A-associated bsAbs may not be effective in these cases (39).
In addition to NK cell-associated bsAbs, several NK cell-associated fusion proteins have been designed as engagers of NK cells and tumor cells. These fusion proteins are combinations of one ligand of an NK cell-associated activating receptor and one TAA-associated antibody fragment or ligand. These fusion proteins are known as bispecific immunoligands. ULBP2-aCEA redirects NK cells to colon carcinoma (169), while rG7S-MICA redirects NK cells to CD24+ HCC cells (170), and both are based on the receptor NKG2D. B7-H6:HER2-scFv and AICL:HER2-scFv are bispecific immunoligands based on NKp30 and NKp80, respectively (171). NKG2D-IL-15 is another bispecific fusion protein, which binds to gastric cancer cells via a fragment of the NKG2D domain and trans-presents IL-15 to NK cells and CD8+ T cells, to induce expansion (172-174).
Combination of NK cell therapy with oncolytic viruses
More than one hundred years ago, physicians found that tumors in patients with cancer would decrease in size after virus infection. This discovery led to current oncolytic virotherapy (175). Natural or genetically modified virus species, which are known as oncolytic viruses can be used to selectively lyse tumor cells. These oncolytic viruses kill tumor cells by directly replicating in and lysing tumor cells or by activating and recruiting immune effectors to the tumor site, or via both mechanisms. They provide a way to overcome several pathways through which tumor cells escape immune surveillance.
Physical tumor barriers represent a critical limitation of solid tumor treatment. However, oncolytic virus vectors may penetrate a tumor, replicate, and spread inside the tumor. Hence, they may enable immune effectors to exert better antitumor efficacy. The oncolytic viruses that are currently under study include adenovirus (ADV), herpes simplex virus (HSV), reovirus, Newcastle disease virus (NDV), vesicular stomatitis virus (VSV), measles virus, vaccinia virus, parvovirus, Maraba virus, rhabdovirus and influenza A virus (134,176-178). Many of these oncolytic viruses have passed the preclinical study phase and moved into clinical trials. The adenovirus-based therapeutic oncolytic virus H101 was approved in China in 2005. Another oncolytic virus, HSV-1 (also known as T-Vec), armed with granulocyte-macrophage colony-stimulating factor (GM-CSF) was approved by the FDA in 2015.
Both preclinical and clinical studies on several of these oncolytic viruses have shown that NK cells are important immune effectors of pharmacologic mechanisms. The therapeutic effect of localized NDV treatment combined with systemic CTLA-4 blockade therapy is dependent on NK cells, CTL and IFN-γ (179). Depletion of NK cells limits therapeutic outcomes significantly. In the treatment of tumors using HSV-1 (ICP34.5 null), subsequent infiltration of NK cells and CTLs is observed (180). In a phase I clinical trial of oncolytic reovirus treatment, enhancement of circulating NK cells was reported (181). DCs activated by oncolytic reovirus promote the antitumor effects of NK cells (182). IL-28 induced by oncolytic VSV facilitates the activation of NK cells (183).
Oncolytic viruses may induce the expression of pathogen pattern receptors (PPRs) or certain viral ligands of activating NK cell receptors by infected tumor cells. These PPRs and viral ligands could stimulate NK cells by engaging Toll-like receptors and NK cell receptors (NCRs including NKp46, NKp44 and NKp30), respectively (134). For example, HSV directly activates NK cells via TLR2 (184), and influenza virus activates NK cells via NKp44 or NKp46 (185,186). The hemagglutinin neuraminidase protein of paramyxovirus is a viral ligand for NKp46 and NKp44 (178). Virus infection also results in “missing-self” signals (e.g., reduced expression of MHC class I for KIRs) and “stressed” signals (e.g., increased expression of MICA for NKG2D or PVR for DNAM-1) on infected tumor cells, which may disrupt several of the evasion strategies of tumor cells. Thus, the antitumor effects of NK cells are enhanced by oncolytic virotherapy.
Furthermore, additional therapeutic genes have been added to oncolytic virus vectors with the goal of enhancing the oncolytic effect. The most effective example is T-Vec, which is armed with GM-CSF. These additional armed genes serve several therapeutic roles: disruption of the physical tumor barrier, modulation of host immunity, and local expression of therapeutic antibodies. This design has been realized in recent research, and they may help to break the tumor barriers. VCN-01 is a human ADV-based oncolytic virus vector armed with PH20 hyaluronidase, which degrades hyaluronic acid (major extracellular tumor matrix) (187). The oncolytic viruses may also enhance the recruitment of NK cells. An oncolytic parvovirus armed with CCL7 or IL-2 was shown to enhance the recruitment of NK cells in pancreatic ductal adenocarcinoma (188). The oncolytic Maraba virus armed with IL-12 promotes the recruitment and function of NK cells (176), and oncolytic ADV armed with IL-12 combined with oncolytic ADV armed with TRAIL induced NK cell tumor infiltration in preclinical models of human hepatocellular carcinoma (HCC) (189). And the intratumoral injection of oncolytic influenza A virus armed with IL-15 increases the tumor infiltration of NK cells and T cells (177). The oncolytic viruses are even further armed with therapeutic antibodies to get more powerful. Full-length trastuzumab has been encoded in an oncolytic ADV vector to produce trastuzumab locally, in order to penetrate the physical tumor barrier and decrease the administration dose. NK cell accumulation has been observed in tumor-draining lymph nodes (190).
Oncolytic virotherapy is also effective for treating hematopoietic malignant disease besides solid tumors. Cytomegalovirus (CMV) reactivation in bone marrow transplantation patients has been associated with anti-leukemic effects and better clinical outcomes (134). UV light-inactivated HSV-1 promotes NK cell activation, migration, degranulation and cytokine production, which potently induces cytolysis of leukemic cells (191). This finding implies that pre-stimulation of donor PBMCs by UV-inactivated HSV-1 ex vivo before infusion might enhance therapeutic efficacy.
Although oncolytic virotherapy was introduced with promising therapeutic advantages, there is one substantial limitation to this approach. In some cases, premature clearance of an oncolytic virus by the host immune system occurs before the therapeutic effect is established. NK cells always play a critical role in this clearance. Pre-storage of both neutralizing antibodies and innate immune cells will limit the spread of the virus before adaptive immunity is functional in the early stages of viral infection. This negative effect limits replication and spreading within the tumor when low doses of an oncolytic virus are administered. Hence, a few questions should first be considered: 1) How do NK cells respond to virus challenge dynamically? One clinical trial has provided an example, where reovirus delivered intravenously was found to be undetectable in the blood one hour after infection (192). Another study revealed a wider range of the activation kinetics of NK cells post-infection under controlled conditions (193); 2) Does the viral vector species initiate the memory-like NK cell responses? This concept is novel but has been verified in studies of CMV infection. Certain NK cell subsets with particular activating receptors could generate a stronger response against subsequent exposure to a once-administered CMV (194); 3) The diversity of the individual genetic complement of KIRs may improve antivirus immunity, as shown in several cases of HCV infection; and 4) The characteristics of certain indications should also be considered.
A perfect resolution to this problem might not exist in this complicated context; however, a few hints may be helpful. For those viral vectors whose therapeutic effect is independent of NK cells, strategies to suppress NK cells should be considered; however, for those viral vectors whose therapeutic effect depends on NK cells, suppressive approaches should be temporary at the early stage, without harming the antiviral function of NK cells. And the activation kinetics of NK cells should be considered for later stages. For example, pretreatment with a single dose of TGF-β in glioblastoma-bearing mice before oncolytic HSV (oHSV) administration has been shown to provide a temporary immunosuppressive window for the replication of oHSV, with the aim of acquiring the maximal therapeutic benefits (195). However, it has not been shown whether NK cells contribute to antitumor effects at the later stage. Neddylation is required for the production of IFN-β during the early phase of HSV-1 infection. MLN4924, a chemotherapeutic agent being studied in clinical trials, can be employed to decrease the production of IFN-β in the early phase by inhibiting neddylation without prolonged effects (196).
Several other interesting combined therapies have been reported. One group found that combined treatment with bortezomib and oncolytic HSV-1 enhances the NK cell antitumor effects against glioma, and this combined therapy in conjunction with NK cell adoptive transfer therapy prolongs the survival of glioma-bearing mice significantly (197). Another approach using oncolytic HSV-1 in combination with EGFR-CAR-NK-92 cells resulted in better antitumor effects and significantly longer survival of tumor-bearing mice (198). It has also been reported that combined therapy involving oncolytic reovirus and rituximab activates NK cells via IFN-α and enhances the ADCC effect (199).
Thus, a synergistic approach involving oncolytic virotherapy and NK cells provides a promising combined cancer therapy strategy.
Conclusions
The restoration of NK cell antitumor activity has long been a desirable approach for controlling cancer growth. Recent studies of NK cell biology as well as NK cell-based translational medicine have shown a great potential for NK cells in tumor control. We have reviewed some of the research in this exciting field, with the intent of inspiring more novel ideas. Adoptive cellular immunotherapy, genetic modification and combination therapy with therapeutic antibodies, bispecific proteins, and oncolytic virotherapy may result in additional clinical benefits in the future. The basic understanding of the receptors of NK cells, subpopulations of NK cells, tissue-specific NK cells and memory-like NK cells should be considered in the future study of NK cell immunotherapy. The complicated heterogeneity of NK cells may influence the efficacy and safety of NK cell-based immunotherapy. Tissue-specific NK cells may also be important for the selection of clinical indications. We have already learned a lot about NK cells as powerful components to defeat cancer; however, they are still mysterious as to our current knowledge. The more we learn about their biology, the better we can employ them to fight cancer. It is expectable that NK cell malfunctions in tumor patients will be classified and fixed precisely with our growing knowledge in the future.
Acknowledgements
This work was supported by the Natural Science Foundation of China (No. 91429303, 31390433, 91542000, 91542114 and 31570893), and the Ministry of Science & Technology of China (973 Basic Science Project 2013CB944902 and 2013CB530506).
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Vivier E, Tomasello E, Baratin M, et al. Functions of natural killer cells. Nat Immunol 2008;9:503–10. DOI:10.1038/ni1582
- Vivier E, Raulet DH, Moretta A, et al. Innate or adaptive immunity? The example of natural killer cells. Science 2011;331:44–9.
- Averdam A, Petersen B, Rosner C, et al. A novel system of polymorphic and diverse NK cell receptors in primates. PLoS Genet 2009;5:e1000688. DOI:10.1371/journal.pgen.1000688
- Vivier E, Nunès JA, Vély F. Natural killer cell signaling pathways. Science 2004;306:1517–9. DOI:10.1126/science.1103478
- Long EO, Kim HS, Liu D, et al. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol 2013;31:227–58. DOI:10.1146/annurev-immunol-020711-075005
- Campbell KS, Purdy AK. Structure/function of human killer cell immunoglobulin-like receptors: lessons from polymorphisms, evolution, crystal structures and mutations. Immunology 2011;132:315–25. DOI:10.1111/j.1365-2567.2010.03398.x
- Thielens A, Vivier E, Romagné F. NK cell MHC class I specific receptors (KIR): from biology to clinical intervention. Curr Opin Immunol 2012;24:239–45. DOI:10.1016/j.coi.2012.01.001
- Zhang J, Basher F, Wu JD. NKG2D ligands in tumor immunity: two sides of a coin. Front Immunol 2015;6:97. DOI:10.3389/fimmu.2015.00097
- Wieten L, Mahaweni NM, Voorter CE, et al. Clinical and immunological significance of HLA-E in stem cell transplantation and cancer. Tissue Antigens 2014;84:523–35. [PubMed] DOI:10.1111/tan.12478
- Kruse PH, Matta J, Ugolini S, et al. Natural cytotoxicity receptors and their ligands. Immunol Cell Biol 2014;92:221–9. [PubMed] DOI:10.1038/icb.2013.98
- Stein N, Tsukerman P, Mandelboim O. The paired receptors TIGIT and DNAM-1 as targets for therapeutic antibodies. Hum Antibodies 2017;25:111–9. [PubMed] DOI:10.3233/HAB-160307
- Ochoa MC, Minute L, Rodriguez I, et al. Antibody-dependent cell cytotoxicity: immunotherapy strategies enhancing effector NK cells. Immunology Cell Biol 2017;95:347–55. [PubMed] DOI:10.1038/icb.2017.6
- Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol 2015;15:388–400. [PubMed] DOI:10.1038/nri3839
- Takeda K, Hayakawa Y, Smyth MJ, et al. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med 2001;7:94–100. [PubMed] DOI:10.1038/83416
- Fauriat C, Long EO, Ljunggren HG, et al. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood 2010;115:2167–76. [PubMed] DOI:10.1182/blood-2009-08-238469
- Caligiuri MA. Human natural killer cells. Blood 2008;112:461–9. [PubMed] DOI:10.1182/blood-2007-09-077438
- Wong JL, Muthuswamy R, Bartlett DL, et al. IL-18-based combinatorial adjuvants promote the intranodal production of CCL19 by NK cells and dendritic cells of cancer patients. Oncoimmunology 2013;2:e26245. [PubMed] DOI:10.4161/onci.26245
- Kalinski P, Giermasz A, Nakamura Y, et al. Helper role of NK cells during the induction of anticancer responses by dendritic cells. Mol Immunol 2005;42:535–9. [PubMed] DOI:10.1016/j.molimm.2004.07.038
- Bouwer AL, Saunderson SC, Caldwell FJ, et al. NK cells are required for dendritic cell-based immunotherapy at the time of tumor challenge. J Immunol 2014;192:2514–21. [PubMed] DOI:10.4049/jimmunol.1202797
- Nguyen-Pham TN, Yang DH, Nguyen TA, et al. Optimal culture conditions for the generation of natural killer cell-induced dendritic cells for cancer immunotherapy. Cell Mol Immunol 2012;9:45–53. [PubMed] DOI:10.1038/cmi.2011.23
- Lam VC, Lanier LL. NK cells in host responses to viral infections. Curr Opin Immunol 2017;44:43–51. [PubMed] DOI:10.1016/j.coi.2016.11.003
- Jiang X, Chen Y, Peng H, et al. Memory NK cells: why do they reside in the liver? Cell Mol Immunol 2013;10:196–201. [PubMed] DOI:10.1038/cmi.2013.8
- Tao Y, Li YH, Piao HL, et al. CD56brightCD25+ NK cells are preferentially recruited to the maternal/fetal interface in early human pregnancy . Cell Mol Immunol 2015;12:77–86. [PubMed] DOI:10.1038/cmi.2014.26
- Peng H, Wisse E, Tian Z. Liver natural killer cells: subsets and roles in liver immunity. Cell Mol Immunol 2016;13:328–36. [PubMed] DOI:10.1038/cmi.2015.96
- Vallentin B, Barlogis V, Piperoglou C, et al. Innate lymphoid cells in cancer. Cancer Immunol Res 2015;3:1109–14. [PubMed] DOI:10.1158/2326-6066.CIR-15-0222
- Dewan MZ, Terunuma H, Takada M, et al. Role of natural killer cells in hormone-independent rapid tumor formation and spontaneous metastasis of breast cancer cells in vivo . Breast Cancer Res Treat 2007;104:267–75. [PubMed] DOI:10.1007/s10549-006-9416-4
- Spear P, Wu MR, Sentman ML, et al. NKG2D ligands as therapeutic targets. Cancer Immun 2013;13:8. [PubMed]
- Koch J, Steinle A, Watzl C, et al. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol 2013;34:182–91. [PubMed] DOI:10.1016/j.it.2013.01.003
- Nausch N, Cerwenka A. NKG2D ligands in tumor immunity. Oncogene 2008;27:5944–58. [PubMed] DOI:10.1038/onc.2008.272
- Seidel UJ, Schlegel P, Lang P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front Immunol 2013;4:76. [PubMed] DOI:10.3389/fimmu.2013.00076
- Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer 2016;16:7–19. [PubMed] DOI:10.1038/nrc.2015.5
- Camous X, Pera A, Solana R, et al. NK cells in healthy aging and age-associated diseases. J Biomed Biotechnol 2012;2012:195956. [PubMed] DOI:10.1155/2012/195956
- Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor micro-environment. Nat Immunol 2013;14:1014–22. [PubMed] DOI:10.1038/ni.2703
- Imai K, Matsuyama S, Miyake S, et al. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet 2000;356:1795–9. [PubMed] DOI:10.1016/S0140-6736(00)03231-1
- Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol 2013;132:515-25; quiz 526.
- Zhang QF, Yin WW, Xia Y, et al. Liver-infiltrating CD11b-CD27- NK subsets account for NK-cell dysfunction in patients with hepatocellular carcinoma and are associated with tumor progression. Cell Mol Immunol 2017;14:819–29. [PubMed] DOI:10.1038/cmi.2016.28
- Sun C, Sun H, Zhang C, et al. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol Immunol 2015;12:292–302. [PubMed] DOI:10.1038/cmi.2014.91
- Farnault L, Sanchez C, Baier C, et al. Hematological malignancies escape from NK cell innate immune surveillance: mechanisms and therapeutic implications. Clin Dev Immunol 2012;2012:421702. [PubMed] DOI:10.1155/2012/421702
- Platonova S, Cherfils-Vicini J, Damotte D, et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res 2011;71:5412–22. [PubMed] DOI:10.1158/0008-5472.CAN-10-4179
- Dewan MZ, Takada M, Terunuma H, et al. Natural killer activity of peripheral-blood mononuclear cells in breast cancer patients. Biomed Pharmacother 2009;63:703–6. [PubMed] DOI:10.1016/j.biopha.2009.02.003
- Krneta T, Gillgrass A, Chew M, et al. The breast tumor microenvironment alters the phenotype and function of natural killer cells. Cell Mol Immunol 2016;13:628–39. [PubMed] DOI:10.1038/cmi.2015.42
- Parisi L, Bassani B, Tremolati M, et al. Natural killer cells in the orchestration of chronic inflammatory diseases. J Immunol Res 2017;2017:4218254. [PubMed] DOI:10.1155/2017/4218254
- Wu JD, Higgins LM, Steinle A, et al. Prevalent expression of the immunostimulatory MHC class I chain-related molecule is counteracted by shedding in prostate cancer. J Clin Invest 2004;114:560–8. [PubMed] DOI:10.1172/JCI22206
- Campoli M, Ferrone S. Tumor escape mechanisms: potential role of soluble HLA antigens and NK cells activating ligands. Tissue Antigens 2008;72:321–34. [PubMed] DOI:10.1111/j.1399-0039.2008.01106.x
- Ren J, Nie Y, Lv M, et al. Estrogen upregulates MICA/B expression in human non-small cell lung cancer through the regulation of ADAM17. Cell Mol Immunol 2015;12:768–76. [PubMed] DOI:10.1038/cmi.2014.101
- Battella S, Cox MC, Santoni A, et al. Natural killer (NK) cells and anti-tumor therapeutic mAb: unexplored interactions. J Leukoc Biol 2016;99:87–96. [PubMed] DOI:10.1189/jlb.5VMR0415-141R
- Sanchez-Correa B, Gayoso I, Bergua JM, et al. Decreased expression of DNAM-1 on NK cells from acute myeloid leukemia patients. Immunol Cell Biol 2012;90:109–15. [PubMed] DOI:10.1038/icb.2011.15
- Kearney CJ, Ramsbottom KM, Voskoboinik I, et al. Loss of DNAM-1 ligand expression by acute myeloid leukemia cells renders them resistant to NK cell killing. Oncoimmunology 2016;5:e1196308. [PubMed] DOI:10.1080/2162402X.2016.1196308
- Elkabets M, Ribeiro VS, Dinarello CA, et al. IL-1β regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol 2010;40:3347–57. [PubMed] DOI:10.1002/eji.201041037
- Yang F, Wei Y, Cai Z, et al. Activated cytotoxic lymphocytes promote tumor progression by increasing the ability of 3LL tumor cells to mediate MDSC chemoattraction via Fas signaling. Cell Mol Immunol 2015;12:66–76. [PubMed] DOI:10.1038/cmi.2014.21
- Quatromoni JG, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res 2012;4:376–89. [PubMed]
- Chretien AS, Le Roy A, Vey N, et al. Cancer-induced alterations of NK-mediated target recognition: current and investigational pharmacological strategies aiming at restoring NK-mediated anti-tumor activity. Front Immunol 2014;5:122. [PubMed] DOI:10.3389/fimmu.2014.00122
- Robson NC, Wei H, McAlpine T, et al. Activin-A attenuates several human natural killer cell functions. Blood 2009;113:3218–25. [PubMed] DOI:10.1182/blood-2008-07-166926
- Tufa DM, Ahmad F, Chatterjee D, et al. IL-1β limits the extent of human 6-sulfo LacNAc dendritic cell (slanDC)-mediated NK cell activation and regulates CD95-induced apoptosis. Cell Mol Immunol 2017;14:976–85. [PubMed] DOI:10.1038/cmi.2016.17
- Sitrin J, Ring A, Garcia KC, et al. Regulatory T cells control NK cells in an insulitic lesion by depriving them of IL-2. J Exp Med 2013;210:1153–65. [PubMed] DOI:10.1084/jem.20122248
- Cao X, Cai SF, Fehniger TA, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 2007;27:635–46. [PubMed] DOI:10.1016/j.immuni.2007.08.014
- Allard B, Longhi MS, Robson SC, et al. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev 2017;276:121–44. [PubMed] DOI:10.1111/imr.12528
- Li T, Yang Y, Hua X, et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett 2012;318:154–61. [PubMed] DOI:10.1016/j.canlet.2011.12.020
- Hoskin DW, Mader JS, Furlong SJ, et al. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int J Oncol 2008;32:527–35. [PubMed]
- Ban Y, Zhao Y, Liu F, et al. Effect of indoleamine 2,3-dioxygenase expressed in HTR-8/SVneo cells on decidual NK cell cytotoxicity. Am J Reprod Immunol 2016;75:519–28. [PubMed] DOI:10.1111/aji.12481
- Cheng M, Chen Y, Xiao W, et al. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol 2013;10:230–52. [PubMed] DOI:10.1038/cmi.2013.10
- Miller JS, Soignier Y, Panoskaltsis-Mortari A, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer . Blood 2005;105:3051–7. [PubMed] DOI:10.1182/blood-2004-07-2974
- Suck G, Odendahl M, Nowakowska P, et al. NK-92: an ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol Immunother 2016;65:485–92. [PubMed] DOI:10.1007/s00262-015-1761-x
- Cheng M, Ma J, Chen Y, et al. Establishment, characterization, and successful adaptive therapy against human tumors of NKG cell, a new human NK cell line. Cell transplant 2011;20:1731–46. [PubMed] DOI:10.3727/096368911X580536
- Klingemann HG. Cellular therapy of cancer with natural killer cells-where do we stand? Cytotherapy 2013;15:1185–94. [PubMed] DOI:10.1016/j.jcyt.2013.03.011
- Nagamura-Inoue T, Mori Y, Yizhou Z, et al. Differential expansion of umbilical cord blood mononuclear cell-derived natural killer cells dependent on the dose of interleukin-15 with Flt3L. Exp Hematol 2004;32:202–9. [PubMed] DOI:10.1016/j.exphem.2003.10.013
- Vasu S, Berg M, Davidson-Moncada J, et al. A novel method to expand large numbers of CD56+ natural killer cells from a minute fraction of selectively accessed cryopreserved cord blood for immunotherapy after transplantation . Cytotherapy 2015;17:1582–93. [PubMed] DOI:10.1016/j.jcyt.2015.07.020
- Kao IT, Yao CL, Kong ZL, et al. Generation of natural killer cells from serum-free, expanded human umbilical cord blood CD34+ cells. Stem cells Dev 2007;16:1043–51. [PubMed] DOI:10.1089/scd.2007.0033
-
Spanholtz J, Preijers F, Tordoir M, et al. Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. PloS One 2011;6:e20740.
[PubMed]
DOI:10.1371/journal.pone.0020740>.
- Fujisaki H, Kakuda H, Shimasaki N, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res 2009;69:4010–7. [PubMed] DOI:10.1158/0008-5472.CAN-08-3712
- Deng X, Terunuma H, Nieda M, et al. Synergistic cytotoxicity of ex vivo expanded natural killer cells in combination with monoclonal antibody drugs against cancer cells . Int Immunopharmacol 2012;14:593–605. [PubMed] DOI:10.1016/j.intimp.2012.09.014
- Masuyama J, Murakami T, Iwamoto S, et al. Ex vivo expansion of natural killer cells from human peripheral blood mononuclear cells co-stimulated with anti-CD3 and anti-CD52 monoclonal antibodies . Cytotherapy 2016;18:80–90. [PubMed] DOI:10.1016/j.jcyt.2015.09.011
- Tonn T, Schwabe D, Klingemann HG, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013;15:1563–70. [PubMed] DOI:10.1016/j.jcyt.2013.06.017
- Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology 2014;3:e28147. [PubMed] DOI:10.4161/onci.28147
- Boissel L, Betancur-Boissel M, Lu W, et al. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology 2013;2:e26527. [PubMed] DOI:10.4161/onci.26527
- Jochems C, Hodge JW, Fantini M, et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget 2016;7:86359–73. [PubMed] DOI:10.18632/oncotarget.13411
- Binyamin L, Alpaugh RK, Hughes TL, et al. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol 2008;180:6392–401. [PubMed]
- Roulot A, Héquet D, Guinebretière JM, et al. Tumoral heterogeneity of breast cancer. Ann Biol Clin (Paris) 2016;74:653–60. [PubMed] DOI:10.1684/abc.2016.1192
- Ibragimova MK, Tsyganov MM, Litviakov NV. Natural and chemotherapy-induced clonal evolution of tumors. Biochemistry (Mosc) 2017;82:413–25. [PubMed] DOI:10.1134/S0006297917040022
- Rybinski B, Yun K. Addressing intra-tumoral heterogeneity and therapy resistance. Oncotarget 2016;7:72322–42. [PubMed] DOI:10.18632/oncotarget.11875
- Romanski A, Uherek C, Bug G, et al. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J Cell Mol Med 2016;20:1287–94. [PubMed] DOI:10.1111/jcmm.12810
- Müller T, Uherek C, Maki G, et al. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol Immunother 2008;57:411–23. [PubMed] DOI:10.1007/s00262-007-0383-3
- Esser R, Müller T, Stefes D, et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med 2012;16:569–81. [PubMed] DOI:10.1111/j.1582-4934.2011.01343.x
- Schönfeld K, Sahm C, Zhang C, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther 2015;23:330–8. [PubMed] DOI:10.1038/mt.2014.219
- Han J, Chu J, Keung Chan W, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep 2015;5:11483. [PubMed] DOI:10.1038/srep11483
- Sahm C, Schönfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother 2012;61:1451–61. [PubMed] DOI:10.1007/s00262-012-1212-x
- Jiang H, Zhang W, Shang P, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol 2014;8:297–310. [PubMed] DOI:10.1016/j.molonc.2013.12.001
- Schirrmann T, Pecher G. Human natural killer cell line modified with a chimeric immunoglobulin T-cell receptor gene leads to tumor growth inhibition in vivo . Cancer Gene Ther 2002;9:390–8. [PubMed] DOI:10.1038/sj.cgt.7700453
- Schirrmann T, Pecher G. Specific targeting of CD33+ leukemia cells by a natural killer cell line modified with a chimeric receptor . Leuk Res 2005;29:301–6. [PubMed] DOI:10.1016/j.leukres.2004.07.005
- Chu J, Deng Y, Benson DM, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma . Leukemia 2014;28:917–27. [PubMed] DOI:10.1038/leu.2013.279
- Suerth JD, Morgan MA, Kloess S, et al. Efficient generation of gene-modified human natural killer cells via alpharetroviral vectors. J Mol Med (Berl) 2016;94:83–93. [PubMed] DOI:10.1007/s00109-015-1327-6
- Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer 2012;12:278–87. [PubMed] DOI:10.1038/nrc3236
- Vacchelli E, Pol J, Bloy N, et al. Trial watch: Tumor-targeting monoclonal antibodies for oncological indications. Oncoimmunology 2015;4:e985940. [PubMed] DOI:10.4161/2162402X.2014.985940
- Sapra P, Shor B. Monoclonal antibody-based therapies in cancer: advances and challenges. Pharmacol Ther 2013;138:452–69. [PubMed] DOI:10.1016/j.pharmthera.2013.03.004
- Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 2015;14:561–84. [PubMed] DOI:10.1038/nrd4591
- Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med 2016;375:1767–78. [PubMed] DOI:10.1056/NEJMra1514296
- Molina A. A decade of rituximab: improving survival outcomes in non-Hodgkin’s lymphoma. Annu Rev Med 2008;59:237–50. [PubMed] DOI:10.1146/annurev.med.59.060906.220345
- Maximiano S, Magalhães P, Guerreiro MP, et al. Trastuzumab in the treatment of breast cancer. BioDrugs 2016;30:75–86. [PubMed] DOI:10.1007/s40259-016-0162-9
- Clynes RA, Towers TL, Presta LG, et al. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets . Nat Med 2000;6:443–6. [PubMed] DOI:10.1038/74704
- Petricevic B, Laengle J, Singer J, et al. Trastuzumab mediates antibody-dependent cell-mediated cytotoxicity and phagocytosis to the same extent in both adjuvant and metastatic HER2/neu breast cancer patients. J Transl Med 2013;11:307. [PubMed] DOI:10.1186/1479-5876-11-307
- Beano A, Signorino E, Evangelista A, et al. Correlation between NK function and response to trastuzumab in metastatic breast cancer patients. J Transl Med 2008;6:25. [PubMed] DOI:10.1186/1479-5876-6-25
- Imai M, Landen C, Ohta R, et al. Complement-mediated mechanisms in anti-GD2 monoclonal antibody therapy of murine metastatic cancer. Cancer Res 2005;65:10562–8. [PubMed] DOI:10.1158/0008-5472.CAN-05-1894
- Robak T, Robak E. New anti-CD20 monoclonal antibodies for the treatment of B-cell lymphoid malignancies. BioDrugs 2011;25:13–25. [PubMed] DOI:10.2165/11539590-000000000-00000
- Capuano C, Pighi C, Molfetta R, et al. Obinutuzumab-mediated high-affinity ligation of FcγRIIIA/CD16 primes NK cells for IFNγ production. Oncoimmunology 2017;6:e1290037. [PubMed] DOI:10.1080/2162402X.2017.1290037
- Awasthi A, Ayello J, Van de Ven C, et al. Obinutuzumab (GA101) compared to rituximab significantly enhances cell death and antibody-dependent cytotoxicity and improves overall survival against CD20+ rituximab-sensitive/-resistant Burkitt lymphoma (BL) and precursor B-acute lymphoblastic leukaemia (pre-B-ALL): potential targeted therapy in patients with poor risk CD20+ BL and pre-B-ALL . Br J Haematol 2015;171:763–75. [PubMed] DOI:10.1111/bjh.13764
- Balasa B, Yun R, Belmar NA, et al. Elotuzumab enhances natural killer cell activation and myeloma cell killing through interleukin-2 and TNF-α pathways. Cancer Immunol Immunother 2015;64:61–73. [PubMed] DOI:10.1007/s00262-014-1610-3
- Spector NL, Blackwell KL. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol 2009;27:5838–47. [PubMed] DOI:10.1200/JCO.2009.22.1507
- Taylor RJ, Chan SL, Wood A, et al. FcγRIIIa polymorphisms and cetuximab induced cytotoxicity in squamous cell carcinoma of the head and neck. Cancer Immunol Immunother 2009;58:997–1006. [PubMed] DOI:10.1007/s00262-008-0613-3
- López-Albaitero A, Lee SC, Morgan S, et al. Role of polymorphic Fc γ receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells . Cancer Immunol Immunother 2009;58:1853–64. [PubMed] DOI:10.1007/s00262-009-0697-4
- Kimura H, Sakai K, Arao T, et al. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor. Cancer Sci 2007;98:1275–80. [PubMed] DOI:10.1111/j.1349-7006.2007.00510.x
- Kurai J, Chikumi H, Hashimoto K, et al. Antibody-dependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines. Clin Cancer Research 2007;13:1552–61. [PubMed] DOI:10.1158/1078-0432.CCR-06-1726
- Collins SM, Bakan CE, Swartzel GD, et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol Immunother 2013;62:1841–9. [PubMed] DOI:10.1007/s00262-013-1493-8
- Wang Y, Sanchez L, Siegel DS, et al. Elotuzumab for the treatment of multiple myeloma. J Hematol Oncol 2016;9:55. [PubMed] DOI:10.1186/s13045-016-0284-z
- Dall’Ozzo S, Tartas S, Paintaud G, et al. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res 2004;64:4664–9. [PubMed] DOI:10.1158/0008-5472.CAN-03-2862
- Di Modica M, Sfondrini L, Regondi V, et al. Taxanes enhance trastuzumab-mediated ADCC on tumor cells through NKG2D-mediated NK cell recognition. Oncotarget 2016;7:255–65. [PubMed] DOI:10.18632/oncotarget.6353
- Kellner C, Hallack D, Glorius P, et al. Fusion proteins between ligands for NKG2D and CD20-directed single-chain variable fragments sensitize lymphoma cells for natural killer cell-mediated lysis and enhance antibody-dependent cellular cytotoxicity. Leukemia 2012;26:830–4. [PubMed] DOI:10.1038/leu.2011.288
- Parsons MS, Richard J, Lee WS, et al. NKG2D acts as a co-receptor for natural killer cell-mediated anti-HIV-1 antibody-dependent cellular cytotoxicity. AIDS Res Hum Retroviruses 2016;32:1089–96. [PubMed] DOI:10.1089/AID.2016.0099
- Inagaki A, Ishida T, Yano H, et al. Expression of the ULBP ligands for NKG2D by B-NHL cells plays an important role in determining their susceptibility to rituximab-induced ADCC. Int J Cancer 2009;125:212–21. [PubMed] DOI:10.1002/ijc.24351
- Ferris RL, Jaffee EM, Ferrone S. Tumor antigen-targeted, monoclonal antibody-based immuno-therapy: clinical response, cellular immunity, and immunoescape. J Clin Oncol 2010;28:4390–9. [PubMed] DOI:10.1200/JCO.2009.27.6360
- Terszowski G, Klein C, Stern M. KIR/HLA interactions negatively affect rituximab- but not GA101 (obinutuzumab)-induced antibody-dependent cellular cytotoxicity. J Immunol 2014;192:5618–24. [PubMed] DOI:10.4049/jimmunol.1400288
- Chan WK, Kung Sutherland M, Li Y, et al. Antibody-dependent cell-mediated cytotoxicity overcomes NK cell resistance in MLL-rearranged leukemia expressing inhibitory KIR ligands but not activating ligands. Clin Cancer Res 2012;18:6296–305. [PubMed] DOI:10.1158/1078-0432.CCR-12-0668
- Capuano C, Romanelli M, Pighi C, et al. Anti-CD20 therapy acts via FcγRIIIA to diminish responsiveness of human natural killer cells. Cancer Res 2015;75:4097–108. [PubMed] DOI:10.1158/0008-5472.CAN-15-0781
- Morales-Estevez C, De la Haba-Rodriguez J, Manzanares-Martin B, et al. KIR genes and their ligands predict the response to anti-EGFR monoclonal antibodies in solid tumors. Front Immunol 2016;7:561. [PubMed] DOI:10.3389/fimmu.2016.00561
- Romagné F, André P, Spee P, et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood 2009;114:2667–77. [PubMed] DOI:10.1182/blood-2009-02-206532
- Vey N, Bourhis JH, Boissel N, et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood 2012;120:4317–23. [PubMed] DOI:10.1182/blood-2012-06-437558
- Kohrt HE, Thielens A, Marabelle A, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood 2014;123:678–86. [PubMed] DOI:10.1182/blood-2013-08-519199
- Benson DM Jr, Hofmeister CC, Padmanabhan S, et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood 2012;120:4324–33. [PubMed] DOI:10.1182/blood-2012-06-438028
- Benson DM Jr, Cohen AD, Jagannath S, et al. A phase I trial of the anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin Cancer Res 2015;21:4055–61. [PubMed] DOI:10.1158/1078-0432.CCR-15-0304
- Nijhof IS, Lammerts van Bueren JJ, van Kessel B, et al. Daratumumab-mediated lysis of primary multiple myeloma cells is enhanced in combination with the human anti-KIR antibody IPH2102 and lenalidomide. Haematologica 2015;100:263–8. [PubMed] DOI:10.3324/haematol.2014.117531
- Kochan G, Escors D, Breckpot K, et al. Role of non-classical MHC class I molecules in cancer immuno-suppression. Oncoimmunology 2013;2:e26491. [PubMed] DOI:10.4161/onci.26491
- González A, Rebmann V, LeMaoult J, et al. The immunosuppressive molecule HLA-G and its clinical implications. Criti Rev Clin Lab Sci 2012;49:63–84. [PubMed] DOI:10.3109/10408363.2012.677947
- Heidenreich S, Zu Eulenburg C, Hildebrandt Y, et al. Impact of the NK cell receptor LIR-1 (ILT-2/CD85j/LILRB1) on cytotoxicity against multiple myeloma. Clin Dev Immunol 2012;2012:652130. [PubMed] DOI:10.1155/2012/652130
- Godal R, Bachanova V, Gleason M, et al. Natural killer cell killing of acute myelogenous leukemia and acute lymphoblastic leukemia blasts by killer cell immunoglobulin-like receptor-negative natural killer cells after NKG2A and LIR-1 blockade. Biol Blood Marrow Transplant 2010;16:612–21. [PubMed] DOI:10.1016/j.bbmt.2010.01.019
- Cantoni C, Grauwet K, Pietra G, et al. Role of NK cells in immunotherapy and virotherapy of solid tumors. Immunotherapy 2015;7:861–82. [PubMed] DOI:10.2217/imt.15.53
- Johnston RJ, Comps-Agrar L, Hackney J, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function . Cancer Cell 2014;26:923–37. [PubMed] DOI:10.1016/j.ccell.2014.10.018
- Groh V, Wu J, Yee C, et al. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002;419:734–8. [PubMed] DOI:10.1038/nature01112
- Bastid J, Regairaz A, Bonnefoy N, et al. Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res 2015;3:254–65. [PubMed] DOI:10.1158/2326-6066.CIR-14-0018
- Raskovalova T, Huang X, Sitkovsky M, et al. Gs protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J Immunol 2005;175:4383–91. [PubMed]
- Gao ZW, Dong K, Zhang HZ. The roles of CD73 in cancer. Biomed Res Int 2014;2014:460654. [PubMed] DOI:10.1155/2014/460654
- Häusler SF, Del Barrio IM, Diessner J, et al. Anti-CD39 and anti-CD73 antibodies A1 and 7G2 improve targeted therapy in ovarian cancer by blocking adenosine-dependent immune evasion. Am J Transl Res 2014;6:129–39. [PubMed]
- Becknell B, Caligiuri MA. Interleukin-2, interleukin-15, and their roles in human natural killer cells. Adv Immunol 2005;86:209–39. [PubMed] DOI:10.1016/S0065-2776(04)86006-1
- Ni J, Miller M, Stojanovic A, et al. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med 2012;209:2351–65. [PubMed] DOI:10.1084/jem.20120944
- Zundler S, Neurath MF. Interleukin-12: Functional activities and implications for disease. Cytokine Growth Factor Rev 2015;26:559–68. [PubMed] DOI:10.1016/j.cytogfr.2015.07.003
- Wawrocki S, Druszczynska M, Kowalewicz-Kulbat M, et al. Interleukin 18 (IL-18) as a target for immune intervention. Acta biochim Pol 2016;63:59–63. [PubMed] DOI:10.18388/abp.2015_1153
- Collins M, Whitters MJ, Young DA. IL-21 and IL-21 receptor: a new cytokine pathway modulates innate and adaptive immunity. Immunol Res 2003;28:131–40. [PubMed] DOI:10.1385/IR:28:2:131
- Sivori S, Cantoni C, Parolini S, et al. IL-21 induces both rapid maturation of human CD34+ cell precursors towards NK cells and acquisition of surface killer Ig-like receptors. Eur J Immunol 2003;33:3439–47. [PubMed] DOI:10.1002/eji.200324533
- Rosenberg SA, Lotze MT, Muul LM, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med 1985;313:1485–92. [PubMed] DOI:10.1056/NEJM198512053132327
- Nelson BH. IL-2, regulatory T cells, and tolerance. J Immunol 2004;172:3983–8. [PubMed]
- Malek TR. The main function of IL-2 is to promote the development of T regulatory cells. J Leukoc Biol 2003;74:961–5. [PubMed] DOI:10.1189/jlb.0603272
- Felices M, Chu S, Kodal B, et al. IL-15 super-agonist (ALT-803) enhances natural killer (NK) cell function against ovarian cancer. Gynecol Oncol 2017;145:453–61. [PubMed] DOI:10.1016/j.ygyno.2017.02.028
- Vego H, Sand KL, Høglund RA, et al. Monomethyl fumarate augments NK cell lysis of tumor cells through degranulation and the upregulation of NKp46 and CD107a. Cell Mol Immunol 2016;13:57–64. [PubMed] DOI:10.1038/cmi.2014.114
- Bartkowiak T, Curran MA. 4-1BB agonists: multi-potent potentiators of tumor immunity. Front Oncol 2015;5:117. [PubMed] DOI:10.3389/fonc.2015.00117
- Baessler T, Charton JE, Schmiedel BJ, et al. CD137 ligand mediates opposite effects in human and mouse NK cells and impairs NK-cell reactivity against human acute myeloid leukemia cells. Blood 2010;115:3058–69. [PubMed] DOI:10.1182/blood-2009-06-227934
- Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo . Proc Nati Acad Sci U S A 2001;98:11521–6. [PubMed] DOI:10.1073/pnas.201238598
- Diefenbach A, Jensen ER, Jamieson AM, et al. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001;413:165–71. [PubMed] DOI:10.1038/35093109
- Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 2006;355:1018–28. [PubMed] DOI:10.1056/NEJMoa063842
- Reiners KS, Kessler J, Sauer M, et al. Rescue of impaired NK cell activity in hodgkin lymphoma with bispecific antibodies in vitro and in patients . Mol Ther 2013;21:895–903. [PubMed] DOI:10.1038/mt.2013.14
- Schmohl JU, Gleason MK, Dougherty PR, et al. Heterodimeric bispecific single chain variable fragments (scFv) killer engagers (BiKEs) enhance NK-cell activity against CD133+ colorectal cancer cells. Target Oncol 2016;11:353–61. [PubMed] DOI:10.1007/s11523-015-0391-8
- Reusch U, Burkhardt C, Fucek I, et al. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs 2014;6:728–39. [PubMed] DOI:10.4161/mabs.28591
- Gleason MK, Ross JA, Warlick ED, et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets . Blood 2014;123:3016–26. [PubMed] DOI:10.1182/blood-2013-10-533398
- Vallera DA, Zhang B, Gleason MK, et al. Heterodimeric bispecific single-chain variable-fragment antibodies against EpCAM and CD16 induce effective antibody-dependent cellular cytotoxicity against human carcinoma cells. Cancer Biother Radiopharm 2013;28:274–82. [PubMed] DOI:10.1089/cbr.2012.1329
- Turini M, Chames P, Bruhns P, et al. A FcγRIII-engaging bispecific antibody expands the range of HER2-expressing breast tumors eligible to antibody therapy. Oncotarget 2014;5:5304–19. [PubMed] DOI:10.18632/oncotarget.2093
- Dong B, Zhou C, He P, et al. A novel bispecific antibody, BiSS, with potent anti-cancer activities. Cancer Biol Ther 2016;17:364–70. [PubMed] DOI:10.1080/15384047.2016.1139266
- Osaki T, Fujisawa S, Kitaguchi M, et al. Development of a bispecific antibody tetramerized through hetero-associating peptides. FEBS 2015;282:4389–401. [PubMed] DOI:10.1111/febs.13505
- Schmohl JU, Felices M, Taras E, et al. Enhanced ADCC and NK cell activation of an anticarcinoma bispecific antibody by genetic insertion of a modified IL-15 cross-linker. Mol Ther 2016;24:1312–22. [PubMed] DOI:10.1038/mt.2016.88
- Schmohl JU, Felices M, Todhunter D, et al. Tetraspecific scFv construct provides NK cell mediated ADCC and self-sustaining stimuli via insertion of IL-15 as a cross-linker. Oncotarget 2016;7:73830–44. [PubMed] DOI:10.18632/oncotarget.12073
- Wu J, Fu J, Zhang M, et al. AFM13: a first-in-class tetravalent bispecific anti-CD30/CD16A antibody for NK cell-mediated immunotherapy. J Hematol Oncol 2015;8:96. [PubMed] DOI:10.1186/s13045-015-0188-3
- Rothe A, Sasse S, Topp MS, et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood 2015;125:4024–31. [PubMed] DOI:10.1182/blood-2014-12-614636
- Rothe A, Jachimowicz RD, Borchmann S, et al. The bispecific immunoligand ULBP2-aCEA redirects natural killer cells to tumor cells and reveals potent anti-tumor activity against colon carcinoma. Int J Cancer 2014;134:2829–40. [PubMed] DOI:10.1002/ijc.28609
- Wang T, Sun F, Xie W, et al. A bispecific protein rG7S-MICA recruits natural killer cells and enhances NKG2D-mediated immunosurveillance against hepatocellular carcinoma. Cancer Lett 2016;372:166–78. [PubMed] DOI:10.1016/j.canlet.2016.01.001
- Peipp M, Derer S, Lohse S, et al. HER2-specific immunoligands engaging NKp30 or NKp80 trigger NK-cell-mediated lysis of tumor cells and enhance antibody-dependent cell-mediated cytotoxicity. Oncotarget 2015;6:32075–88. [PubMed] DOI:10.18632/oncotarget.5135
- Xia Y, Chen B, Shao X, et al. Treatment with a fusion protein of the extracellular domains of NKG2D to IL-15 retards colon cancer growth in mice. J Immunother 2014;37:257–66. [PubMed] DOI:10.1097/CJI.0000000000000033
- Yan C, Jie L, Yongqi W, et al. Delivery of human NKG2D-IL-15 fusion gene by chitosan nanoparticles to enhance antitumor immunity. Biochem Biophys Res Commun 2015;463:336–43. [PubMed] DOI:10.1016/j.bbrc.2015.05.065
- Chen Y, Chen B, Yang T, et al. Human fused NKG2D-IL-15 protein controls xenografted human gastric cancer through the recruitment and activation of NK cells. Cell Mol Immunol 2017;14:293–307. [PubMed] DOI:10.1038/cmi.2015.81
- Pol J, Bloy N, Obrist F, et al. Trial Watch: Oncolytic viruses for cancer therapy. Oncoimmunology 2014;3:e28694. [PubMed] DOI:10.4161/onci.28694
- Alkayyal AA, Tai LH, Kennedy MA, et al. NK-cell recruitment is necessary for eradication of peritoneal carcinomatosis with an IL12-expressing maraba virus cellular vaccine. Cancer Immunol Res 2017;5:211–21. [PubMed] DOI:10.1158/2326-6066.CIR-16-0162
- Hock K, Laengle J, Kuznetsova I, et al. Oncolytic influenza A virus expressing interleukin-15 decreases tumor growth in vivo . Surgery 2017;161:735–46. [PubMed] DOI:10.1016/j.surg.2016.08.045
- Matveeva OV, Guo ZS, Shabalina SA, et al. Oncolysis by paramyxoviruses: multiple mechanisms contribute to therapeutic efficiency. Mol Ther Oncolytics 2015;2. pii: 15011.
- Zamarin D, Holmgaard RB, Subudhi SK, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med 2014;6:226ra32. [PubMed] DOI:10.1126/scitranslmed.3008095
- Benencia F, Courrèges MC, Conejo-García JR, et al. HSV oncolytic therapy upregulates interferon-inducible chemokines and recruits immune effector cells in ovarian cancer. Mol Ther 2005;12:789–802. [PubMed] DOI:10.1016/j.ymthe.2005.03.026
- White CL, Twigger KR, Vidal L, et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther 2008;15:911–20. [PubMed] DOI:10.1038/gt.2008.21
- Errington F, Steele L, Prestwich R, et al. Reovirus activates human dendritic cells to promote innate antitumor immunity. J Immunol 2008;180:6018–26. [PubMed]
- Wongthida P, Diaz RM, Galivo F, et al. Type III IFN interleukin-28 mediates the antitumor efficacy of oncolytic virus VSV in immune-competent mouse models of cancer. Cancer Res 2010;70:4539–49. [PubMed] DOI:10.1158/0008-5472.CAN-09-4658
- Kim M, Osborne NR, Zeng W, et al. Herpes simplex virus antigens directly activate NK cells via TLR2, thus facilitating their presentation to CD4 T lymphocytes. J Immunol 2012;188:4158–70. [PubMed] DOI:10.4049/jimmunol.1103450
- Mandelboim O, Lieberman N, Lev M, et al. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 2001;409:1055–60. [PubMed] DOI:10.1038/35059110
- Arnon TI, Lev M, Katz G, et al. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur J Immunol 2001;31:2680–9. [PubMed] DOI:10.1002/1521-4141(200109)31:9<2680::AID-IMMU2680>3.0.CO;2A
- Rodríguez-García A, Giménez-Alejandre M, Rojas JJ, et al. Safety and efficacy of VCN-01, an oncolytic adenovirus combining fiber HSG-binding domain replacement with RGD and hyaluronidase expression. Clin Cancer Res 2015;21:1406–18. [PubMed] DOI:10.1158/1078-0432.CCR-14-2213
- Dempe S, Lavie M, Struyf S, et al. Antitumoral activity of parvovirus-mediated IL-2 and MCP-3/CCL7 delivery into human pancreatic cancer: implication of leucocyte recruitment. Cancer Immunol Immunother 2012;61:2113–23. [PubMed] DOI:10.1007/s00262-012-1279-4
- El-Shemi AG, Ashshi AM, Na Y, et al. Combined therapy with oncolytic adenoviruses encoding TRAIL and IL-12 genes markedly suppressed human hepatocellular carcinoma both in vitro and in an orthotopic transplanted mouse model . J Exp Clin Cancer Res 2016;35:74. [PubMed] DOI:10.1186/s13046-016-0353-8
- Liikanen I, Tähtinen S, Guse K, et al. Oncolytic adenovirus expressing monoclonal antibody trastuzumab for treatment of HER2-positive cancer. Mol Cancer Ther 2016;15:2259–69. [PubMed] DOI:10.1158/1535-7163.MCT-15-0819
- Samudio I, Rezvani K, Shaim H, et al. UV-inactivated HSV-1 potently activates NK cell killing of leukemic cells. Blood 2016;127:2575–86. [PubMed] DOI:10.1182/blood-2015-04-639088
- Adair RA, Roulstone V, Scott KJ, et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci Transl Med 2012;4:138ra77.
- El-Sherbiny YM, Holmes TD, Wetherill LF, et al. Controlled infection with a therapeutic virus defines the activation kinetics of human natural killer cells in vivo . Clin Exp Immunol 2015;180:98–107. [PubMed] DOI:10.1111/cei.12562
- Béziat V, Liu LL, Malmberg JA, et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood 2013;121:2678–88. [PubMed] DOI:10.1182/blood-2012-10-459545
- Han J, Chen X, Chu J, et al. TGFβ treatment enhances glioblastoma virotherapy by inhibiting the innate immune response. Cancer Res 2015;75:5273–82. [PubMed] DOI:10.1158/0008-5472.CAN-15-0894
- Zhang X, Ye Z, Pei Y, et al. Neddylation is required for herpes simplex virus type I (HSV-1)-induced early phase interferon-beta production. Cell Mol Immunol 2016;13:578–83. [PubMed] DOI:10.1038/cmi.2015.35
- Yoo JY, Jaime-Ramirez AC, Bolyard C, et al. Bortezomib treatment sensitizes oncolytic HSV-1-treated tumors to NK cell immunotherapy. Clin Cancer Res 2016;22:5265–76. [PubMed] DOI:10.1158/1078-0432.CCR-16-1003
- Chen X, Han J, Chu J, et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 2016;7:27764–77. [PubMed] DOI:10.18632/oncotarget.8526
- Parrish C, Scott GB, Migneco G, et al. Oncolytic reovirus enhances rituximab-mediated antibody-dependent cellular cytotoxicity against chronic lymphocytic leukaemia. Leukemia 2015;29:1799–810. [PubMed] DOI:10.1038/leu.2015.88