
Voltage-gated K+ channels promote BT-474 breast cancer cell migration
Introduction
A number of ion channels have been proposed as biomarkers or functional molecules in breast cancer metastasis. For instance, voltage-gated Na+ channel (VGSC) Nav1.5 is up-regulated in metastatic breast cancer tissue, and has been postulated as a biomarker and therapeutic targets for breast cancer metastasis (1). How exactly Na+ influx leads to metastasis is unclear, but a report has described that the enhanced invasiveness is due to H+ efflux via Na+-H+ exchanger, which is in turn stimulated by Na+ influx via Nav1.5 (2). Another candidate attracting recent attention is the voltage-gated proton channel (Hv1). The latter allows H+ to flow out of the cells, and thus an acidic environment is created to facilitate the actions of several enzymes important in cancer cell invasiveness and migration, such as matrix metalloproteinases (3,4). Novel blockers of Hv1 may be useful in inhibiting cancer metastasis (5).
As Ca2+ is an important stimulator for cell mobility, Ca2+ influx is believed to be one of the essential motor forces in breast cancer migration. Of note, stromal interaction molecule 1 (STIM1) and Orai1, two protein components of store-operated Ca2+ channel, have been implicated in breast cancer migration (6). In addition, transient receptor potential channel M7 (TRPM7), a Ca2+-permeable non-selective cation channel-kinase, has also been implicated in cancer cell adhesion and migration, as high TRPM7 expression is associated with poor prognosis in breast cancer patients (7,8). Further, this molecule is required for metastasis formation in a mouse xenograft model of human breast cancer. Findings suggest that TRPM7 is a component of a mechanosensory complex to drive breast cancer cell metastasis (9,10).
Voltage-gated K+ (Kv) channels open upon depolarization and allow K+ efflux to repolarize excitable cells (11,12). Depending on sequence homology, Kv channels are classified into Kv1 to Kv12 subfamilies with each Kv subfamily having multiple subtype members (13). Kv1.3, Kv4.1, Kv10.1 and Kv11.1 are present in breast cancer cells (14). Of interest, Kv channels have been implicated in breast cancer migration (15). The mechanism is unknown until Hammadi et al. obtained data showing that blocking or silencing hEag1 (Kv10.1) depolarized breast cancer MDA-MB-231 cells, reducing Ca2+ entry (via Orai1-associated channel), and eventually inhibiting cell migration without affecting cell proliferation (16). Thus, hEag1 is essential in maintaining a negative potential favorable for Ca2+ entry, which is important in cell motility. In this report, Kv channel currents were found to be much higher in migratory than non-migratory breast cancer BT474 cells; blockade of Kv currents by tetraethylammonium (TEA) suppressed cell migration. In contrast to the reported case in MDA-MB-231 cells, migratory BT474 cells had more depolarized membrane potential and reduced Ca2+ entry. Alternative models to explain the roles of Kv channels in migration will be discussed.
Materials and methods
Cell culture
BT474 cells were cultured at 37 °C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) and penicillin-streptomycin (100 U/mL, 100 μg/mL) (Invitrogen).
Separation of migratory cells from non-migratory cells
BT474 cells (3×105) were seeded in the upper chambers of the Transwell (Corning 3428, 24 mm) and allowed to migrate through the porous (8 μm) membrane for 3 d. The upper chamber medium was serum-free whilst the lower chambers contained 10% FBS as a chemoattractant (17,18). We then separated the non-migratory cells from the migratory cells in the following manner: non-migratory cells in the upper chamber were trypsinized and seeded on culture plates, while migratory cells trapped in the membrane were trypsinized, detached and seeded on separate cultures plates. The non-migratory cells and migratory cells were then allowed to settle on their culture plates for 5 h and then subject to electrophysiological recording for Kv currents or to microfluorimetric measurements.
Migration assay
BT474 cells (3×105) were seeded on the upper chamber of the Transwell (Corning 3428, 24 mm) and incubated for 3 d in the absence or presence of pharmacological agents. The cells were allowed to migrate through the porous (8 μm) membrane for 3 d. The upper chamber medium was serum-free whilst the lower chambers contained 10% FBS as a chemoattractant (17,18). After 3 d, the upper chambers were washed thoroughly and the cells in the porous membrane were stained with crystal violet. Five random views of each sample were photographed and the number of cells was counted. The number of cells in treatment groups was normalized with those in the control group and expressed as % control.
Electrophysiology
Electrophysiological experiments were performed as previously reported (19). Cells were voltage-clamped in the whole-cell configuration. Thin-walled borosilicate glass tubes (o.d. 1.5 mm, i.d. 1.10 mm, Sutter Instrument, Novato, CA) were pulled with a micropipette puller (P-87, Sutter Instrument), and then heat polished by a microforge (Narishige Instruments, Inc., Sarasota, FL, USA). The pipettes, filled with intracellular solution, containing (mmol/L): 140 KCl, 1 MgCl2, 1 EGTA, 10 HEPES, and 5 MgATP (pH 7.25 adjusted with KOH), had typical resistance of 4−7 MΩ. The bath solution contained (mmol/L): 140 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES (pH 7.4 adjusted with NaOH). The currents were recorded using an EPC-10 amplifier with Pulse 8.60 acquisition software and analyzed by Pulsefit 8.60 software (HEKA Electronik, Lambrecht, Germany). Data were filtered at 2 kHz and sampled at 10 kHz. After a whole-cell configuration was established, the cells were held at −70 mV and subject to various protocols as detailed in the Protocol. All experiments were performed at room temperature (25 °C).
Reverse transcriptase-polymerase chain reaction (RT-PCR)
Total RNA of BT474 cells was isolated by RNA Isolater (Vazyme, Nanjing, China), precipitated with iso-propanol, washed with 70% ethanol and finally dissolved with nucleic acid stabilized solution (Topgen Biotech., Co., Ltd, Taiwan, China). Total RNA was determined by EpochTM Spectrophotometer System (Bio Tek Instruments, Winooski, VT, USA). Genomic DNA was removed using 4× gDNA wiper Mix at 42 °C for 15 min. Reverse transcription of 1 μg of total RNA to cDNA was performed using HiScript II 1st Strand cDNA Synthesis Kit (Vazyme), and cDNA synthesis was performed at 25 °C for 5 min, 50 °C for 15 min and 85 °C for 2 min on ABI PCR system VeritiTM 96-Well Thermal Cycler (Applied Biosystems, Waltham, USA). Qualitative analysis of 12 target genes, KCNA1 (Kv1.1), KCNA2 (Kv1.2), KCNA3 (Kv1.3), KCNA4 (Kv1.4), KCNA5 (Kv1.5), KCNA6 (Kv1.6), KCNA7 (Kv1.7), KCNB1 (Kv2.1), KCNC1 (Kv3.1), KCNC3 (Kv3.3), KCNC4 (Kv3.4), KCND3 (Kv4.3) and internal control GAPDH with custom designed primers (see below; Topgen Biotech., Co., Ltd, Taiwan, China) was performed by 2X Taq Master Mix (Vazyme) using the following program: 95 °C-5 min, 40 cycles for 95 °C-15 s, 60 °C-15 s, 72 °C-15 s and 72 °C-1 min on ABI PCR system VeritiTM 96-Well Thermal Cycler (Applied Biosystems). Size distribution of target gene PCR products was then examined by an MCE-202 MultiNA Microchip Electrophoresis System with DNA-2500 Kit (Shimadzu, Japan).
Specific primer sequences of each target gene are shown below:
Kv1.1-F, GGAGAGGAACAGGCCCAATAC;
Kv1.1-R, CCCTCCTGGATCTCCATGTAATC;
Kv1.2-F,TCTGCAAAGCCAGAAGAGTAAGC;
Kv1.2-R, AGCTGGTGTCTGCCTGTCATC;
Kv1.3-F, TTCTCCAGCGCGGTCTACTT;
Kv1.3-R,TGGTCACTGGGTGCATATCG;
Kv1.4-F, CATTGCGGGTGTCTTAACCA;
Kv1.4-R,CTGACTGCATTCTGCGTTAGCT;
Kv1.5-F,GACCCTGGAGAATGCAGACAGT;
Kv1.5-R, ATAAAGGGACCTCCGCAAGTC;
Kv1.6-F, TCGTCTCCGTGTTGGTCATTC;
Kv1.6-R, GACTCGACTCACACCACCATTG;
Kv1.7-F, GGAGACGCTGTGTATTTGTTGGT;
Kv1.7-R, AGTGCCACAAAGTAGGGAAGGA;
Kv2.1-F,CTGTGGCTGCCAAGATCCTT;
Kv2.1-R,GTCTGTGGACTGGCCGAACT;
Kv3.1-F, CGAGAGATACGGACCCTGCTT;
Kv3.1-R, CCTCTGTCGGCATATACTTAGCAA;
Kv3.3-F, GCTCTTCGAGGACCCCTACTC;
Kv3.3-R,CACCGTCTTGTTGCTAATATGGAT;
Kv3.4-F, CGCTCTTCGAGGATCCCTACT;
Kv3.4-R,GGGTCTCCAGGCAGAAAGTG;
Kv4.3-F,ATCCCTGCCTCGTTTTGGTA;
Kv4.3-R, ACGCCACTCAAGGAGCAGAT;
GAPDH-F, GCACCACCAACTGCTTAGCA;
GAPDH-R, TCTTCTGGGTGGCAGTGATG.
Microfluorimetric measurement of cytosolic Ca2+
Microfluorimetric measurement of cytosolic Ca2+ concentration was performed using fura-2 as a Ca2+-sensitive fluorescent dye as previously reported (20). In brief, cells were incubated with 5 μmol/L fura-2 AM (Invitrogen) for 1 h at 37 °C and then washed in extracellular bath solution which contained (mmol/L): 140 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES (pH 7.4 adjusted with NaOH). When intracellular Ca2+ release was assayed, Ca2+-free solution was used. This Ca2+-free solution was the same as the extracellular bath solution mentioned above except that Ca2+ was omitted and 100 μmol/L EGTA was supplemented. Cells were alternately excited with 340 nm and 380 nm using an optical filter changer (Lambda 10-2, Sutter Instruments, Novato, CA, USA). Emission was collected at 500 nm and images were captured using a CCD camera (CoolSnap HQ2, Photometrics, Tucson, AZ, USA) linked to an inverted Nikon TE 2000-U microscope. Images were analyzed with an MAG Biosystems Software (Sante Fe, MN). All imaging experiments were performed at room temperature (25 °C).
Statistical analysis
Results are expressed as x±sx. Unpaired or paired Student’s t-test was used for two groups. For multiple group comparisons, one-way analysis of variance (ANOVA) with Tukey’s honest significant difference (HSD) post hoc test was used. A value of P<0.05 is considered statistically significant.
Results
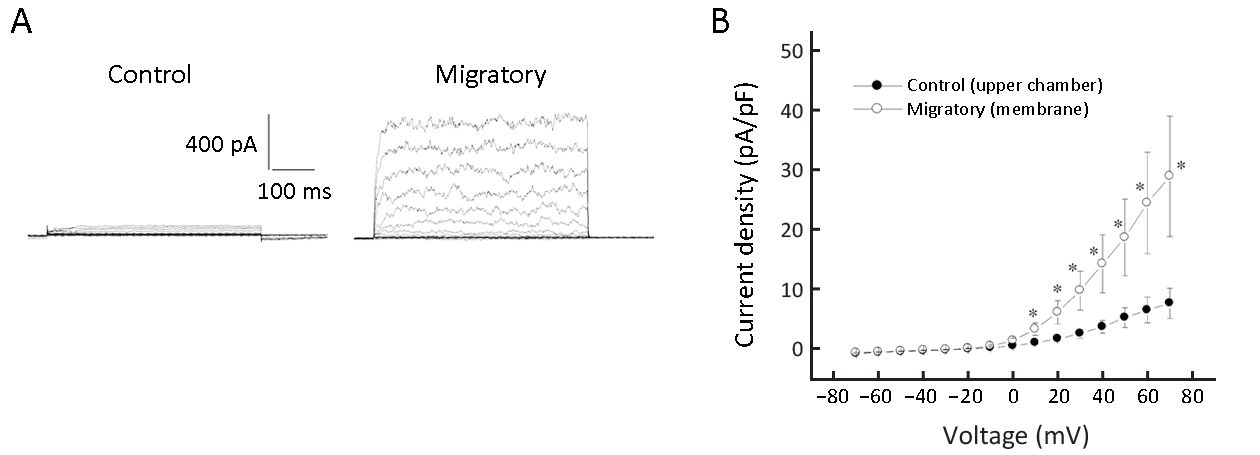
Migratory cells had remarkably increased Kv currents
We observed that only a minor fraction of non-migratory cells (6 out of 36; 16.7%) had Kv currents, while 58.1% of migratory cells (18 out of 31) had Kv currents (albeit of variable size). The current densities were calculated and migratory cells had remarkably larger Kv current density than non-migratory cells (Figure 1). Large-conductance Ca2+-activated K+ (BK) currents are also voltage-activated (12), but we believe the outward K+ currents we observed are Kv currents but not BK currents for dual reasons: first, BK currents are activated by Ca2+ >1 μmol/L, but our pipette solution (hence intracellular milieu) was Ca 2+-free supplemented with 1 mmol/L EGTA. Second, BK currents could be largely inhibited by <1 mmol/L TEA (12), but the currents we observed were only significantly inhibited by >10 mmol/L TEA.
As the lower chambers contained 10% FBS as chemoattractant, we investigated whether the increased expression of Kv current in migratory cells was due to the presence of FBS. Therefore, cells were grown in the absence or presence of 10% FBS for 3 d and then currents were examined. As shown in Figure 2, there was no significant difference in current densities between the two groups, suggesting the up-regulation of Kv currents in migratory cells was not due to stimulation by FBS.

Delayed rectifier type Kv channels were present and were important in cell migration
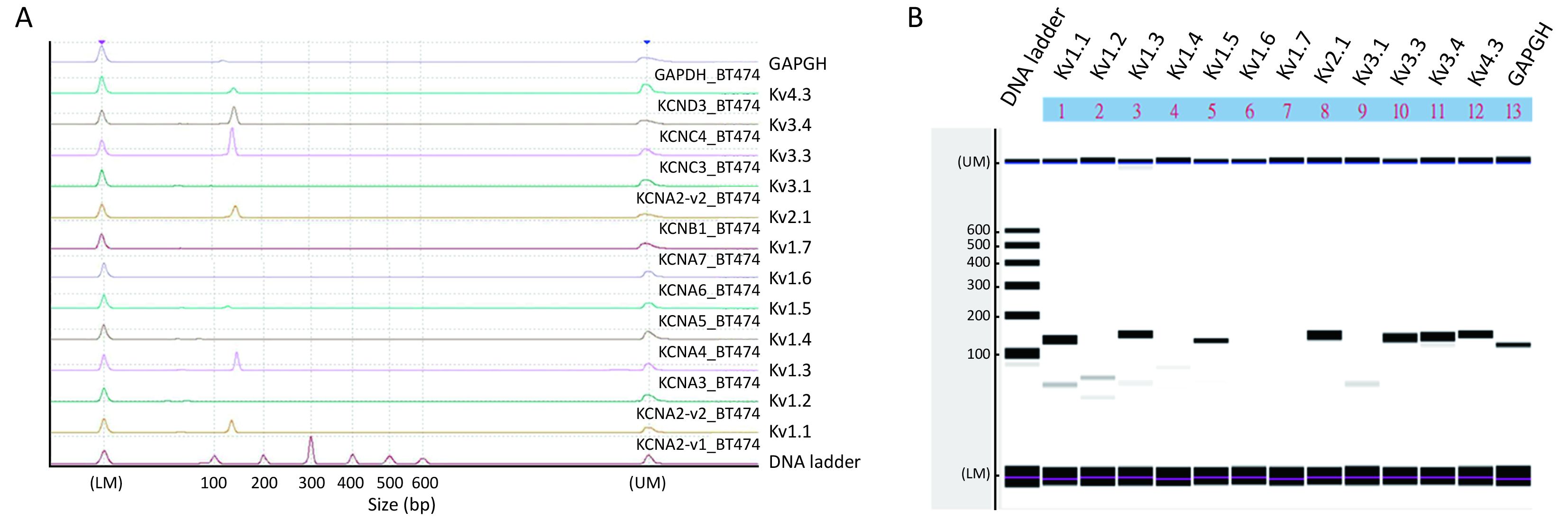
We next examined what Kv channel members were present in BT474 cells using RT-PCR with a microchip electrophoresis system (Figure 3). Electropherogram shows peaks of Kv1.1, Kv1.3, Kv1.5, Kv2.1, Kv3.3, Kv3.4, Kv4.3 and the internal control GAPDH at the expected 100−200 bp range (Figure 3A), which are indicated as major bands in Figure 3B. Very tiny peaks (Figure 3A), indicated as very faint bands in Figure 3B, were non-specific amplifications but were very few. Thus, Kv1.1, Kv1.3, Kv1.5, Kv2.1, Kv3.3, Kv3.4 and Kv4.3 channels were expressed in BT474 cells.
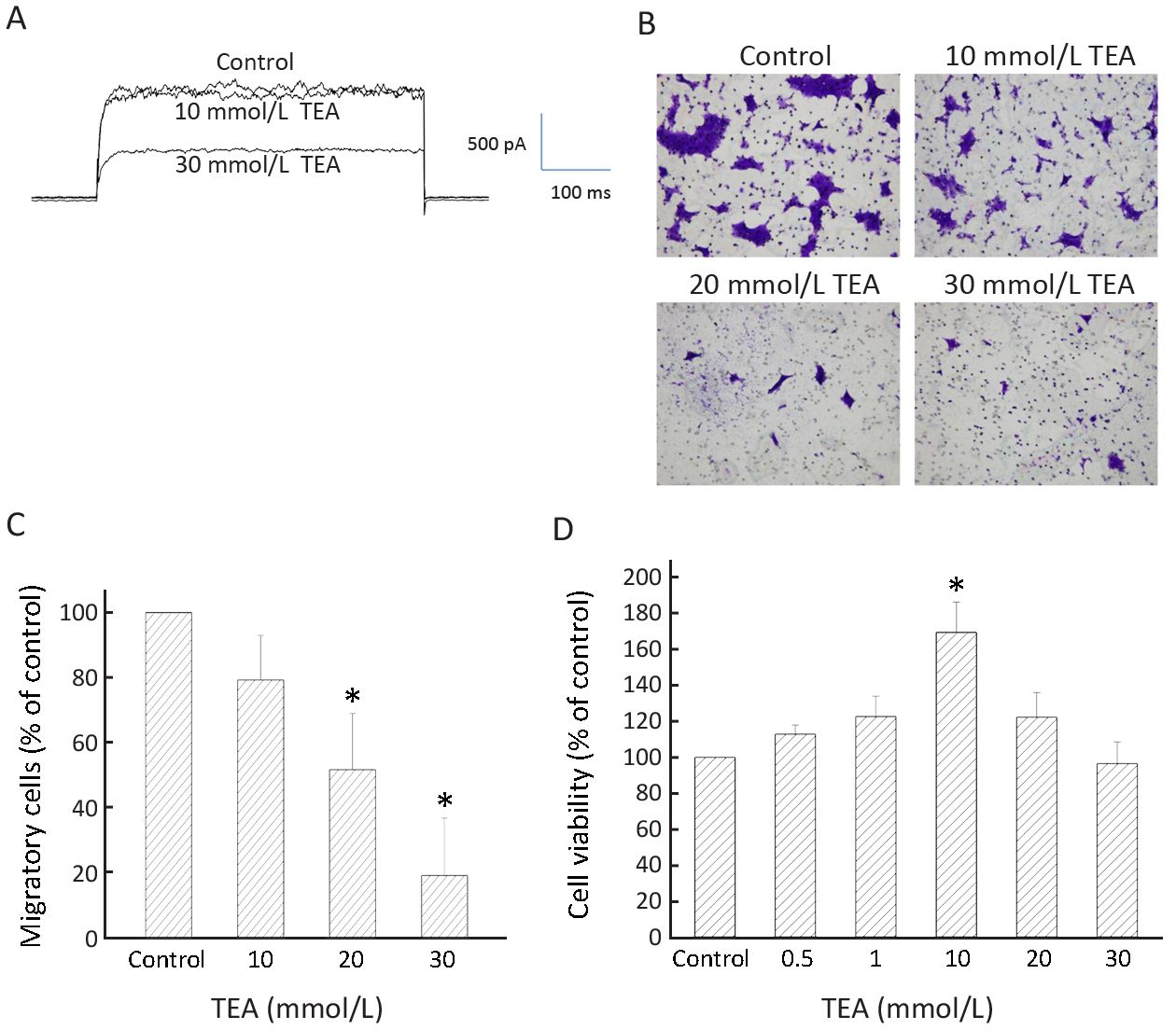
To examine whether the Kv currents played a role in BT474 cell migration, we deployed TEA, a delayed rectifier Kv channel blocker, as a tool to inhibit BT474 cell Kv currents and migration. Significant inhibition of Kv currents required >10 mmol/L TEA (Figure 4A). Consistently, significant inhibition of migration required 20−30 mmol/L TEA (Figure 4B, C). TEA at these concentrations did not affect cell viability when compared to the control which had no TEA (Figure 4D). These results suggest delayed rectifier type Kv currents are important in cell migration. However, 4-aminopyridine (4-AP), an A-type Kv channel blocker, did not inhibit BT474 cell Kv currents and did not significantly affect cell migration (Figure 5). In concordance, BT474 cell Kv currents exhibited very slow inactivation (Figure 1, 4, 5) instead of fast inactivation characteristic of A-type Kv channels.

Migratory cells had more depolarized membrane potential and reduced Ca2+ influx
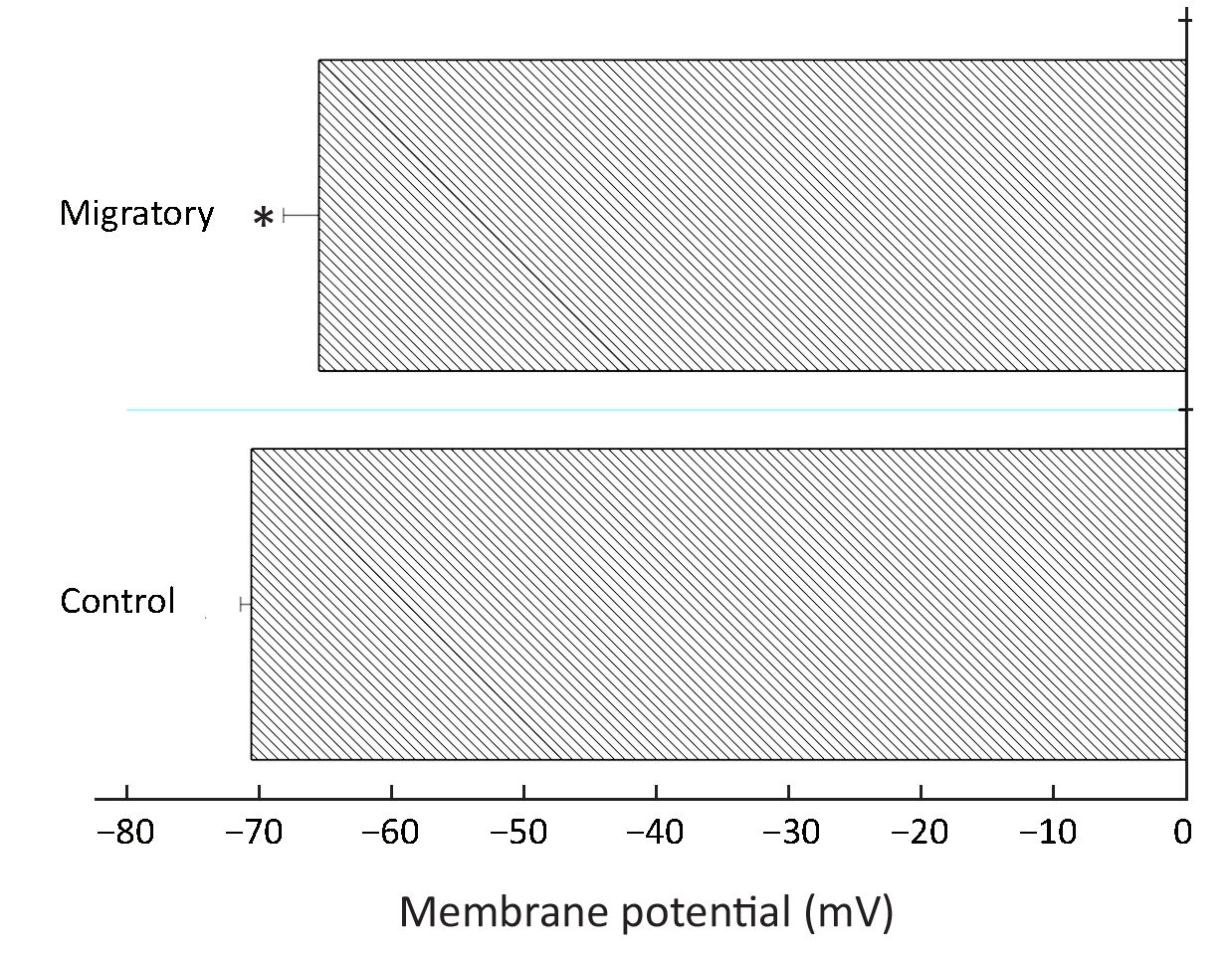
It has been reported before that Kv channels regulate membrane potential which in turn regulates Ca2+ signaling in breast cancer MDA-MB-231 cells (16). More Kv channels may be expected to cause a lower membrane potential (hyperpolarization). Therefore we examined if there was a difference in membrane potential between the non-migratory and migratory BT474 cells. Rather unexpectedly, the migratory cells, having much higher Kv channel density, had slightly but significantly depolarized membrane potential (Figure 6).
We next examined whether there was a difference in Ca2+ signaling between non-migratory and migratory BT474 cells. Cyclopiazonic acid (CPA; intracellular Ca2+ pump inhibitor) was deployed to cause intracellular Ca2+ release, and then replenishment of extracellular Ca2+ resulted in Ca2+ influx. We observed that while intracellular Ca2+ release was similar in the two groups of cells, Ca2+ influx was smaller in the migratory cells (Figure 7). This is consistent with the more depolarized membrane potential in the migratory cells: such depolarization decreased the electric driving force for Ca2+ influx.

Discussion
Data in this report show that there was an increased expression of Kv channels in migratory BT474 cells. In both non-migratory and migratory cells, Kv currents show no or very slow inactivation, suggesting most of the members were of delayed rectifier types. Consistently, TEA (delayed rectifier blocker), but not 4-AP (A-type channel blocker), could significantly inhibit currents and cell migration, suggesting that delayed rectifier Kv channels were important in cell migration. An RT-PCR analysis revealed that several members were present, namely, Kv1.1, Kv1.3, Kv1.5, Kv2.1, Kv3.3, Kv3.4 and Kv4.3.
How do Kv channels promote cell migration? Hammadi et al. obtained data showing that blocking or silencing hEag1 (Kv10.1) depolarized breast cancer MDA-MB-231 cells, reducing Ca2+ entry (via Orai1-associated channel), and eventually inhibiting cell migration without affecting cell proliferation (16). Thus, hEag1 is essential in maintaining a negative membrane potential to be favorable for Ca2+ entry, which is important in cell motility. However, using BT474 cells, our data do not support such a hyperpolarizing role of Kv channels. In fact, migratory cells, which expressed more Kv channels, were more depolarized (Figure 6). Further, migratory cells had reduced Ca2+ entry after store depletion (Figure 7); such reduction is consistent with the more depolarized membrane potential, which decreased the electrical driving force for Ca2+. Therefore, our findings in BT474 cells are novel and significant in showing that Kv channels promote migration via mechanism(s) independent of Ca2+ influx.
In a very recent report, Kv1.1 and 1.3 channels are demonstrated to play a role in MDA-MB-231 cell migration (21). In addition, human ether-à-go-go-related gene 1 (hERG1) plays a role in human osteosarcoma cell migration (22). However, in these two works, electrophysiological measurements and Ca2+ assays were not performed to show whether migratory cells have enhanced K+ currents or altered Ca2+ homeostasis.
Inward rectifier K+ channels (Kir2.2, Kir3.1, Kir6.1, Kir6.2) and Ca2+-activated K+ channels (KCa1.1, KCa2.3 and KCa3.1) are expressed in breast cancer cells (14). Blocking intermediate- and large-conductance KCa channels could inhibit glioma migration and tumor infiltration (23,24). Small- and large-conductance KCa channels are also implicated in breast cancer migration and metastasis; in case of small-conductance KCa channels, they form a complex with Orai-Ca2+ channels so that they could be directly activated by localized Ca2+ elevation with lipid rafts (25). However, a model integrating the roles of Kv, Kir and KCa channels in cancer cell migration is hitherto lacking. Mobile lamellipodia are observed at the leading edge of mobile cells, while the contracting cell body is at the trailing edge. A possible mechanism is proposed in which at the posterior of the moving cell, Kv and KCa channels provide K+ efflux and subsequently water flows out osmotically via aquaporin; this causes local volume decrease at the cell posterior and culminates in rear retraction (14). At the cell anterior, Kir channels provide an influx K+ pathway, osmotically drawing water in via aquaporin; this results in local volume increase and culminates in front protrusion. There is evidence this could provide enough mobile force for cell motility even in the absence of actin polymerization (26). Whether this could occur in breast cancer migration is hitherto unknown and would warrant future investigation.
The non-canonical functions of Kv channels (that is, functions unrelated to ion conduction) in BT474 migration also warrants consideration. In this case, Kv channels may form molecular complexes with other proteins such as integrin to facilitate cell migration. For example, hERG channels have been demonstrated to form complex with vascular endothelial growth factor receptor-1 (VEGFR-1) and β1 integrin to promote the formation of a migratory leukemia phenotype (27).
Besides their most well-known functions in dampening cellular excitability, convincing evidence supports the involvement of Kv channels in cell death, differentiation and functions of a wide variety of cells. For instance, there is an overexpression of Kv channels during apoptosis of neurons, immune cells and muscle cells (28,29). The massive K+ efflux through over-expressed Kv channels leads to loss of intracellular K+ and the reduced [K+]i results in relief of inhibition (thus activation) of pro-apoptotic caspases and nucleases (30,31). K+ efflux through Kv1.1, Kv1.4 and Kv2.1 is responsible for cAMP-stimulated neuritogenesis in mouse neuroblastoma N2A cells (20). Strong evidence from Ilana Lotan’s lab has suggested that, in a manner independent of K+ flow, Kv2.1 directly interacts with syntaxin in facilitating exocytosis, further substantiating the presence of non-canonical functions of Kv channels (32). Kv1.3 has been shown to be involved in breast cancer cell proliferation (33). In this report Kv channels have been further demonstrated to promote breast cancer cell migration. Therefore, there is mounting evidence suggesting Kv channels are versatile molecules which have a variety of functions in addition to provision of K+ efflux for repolarization.
Conclusions
Kv channel expression was up-regulated during BT474 cell migration, and hence Kv channels could be considered as biomarkers or potential therapeutic targets for breast cancer cell migration.
Acknowledgements
The study was funded by the Macau Science and Technology Development Fund (FUNDO PARA O DESENVOLVIMENTO DAS CIÊNCIAS E DA TECNOLOGIA) and the reference number was 002/2015/A1.
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Fraser SP, Diss JK, Chioni AM, et al. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin Cancer Res 2005;11:5381–9. [PubMed] DOI:10.1158/1078-0432.CCR-05-0327
- Brisson L, Gillet L, Calaghan S, et al. NaV1.5 enhances breast cancer cell invasiveness by increasing NHE1-dependent H+ efflux in caveolae . Oncogene 2011;30:2070–6. [PubMed] DOI:10.1038/onc.2010.574
- Wang Y, Li SJ, Pan J, et al. Specific expression of the human voltage-gated proton channel Hv1 in highly metastatic breast cancer cells, promotes tumor progression and metastasis. Biochem Biophys Res Commun 2011;412:353–9. [PubMed] DOI:10.1016/j.bbrc.2011.07.102
- Wang Y, Li SJ, Wu X, et al. Clinicopathological and biological significance of human voltage-gated proton channel Hv1 protein overexpression in breast cancer. J Biol Chem 2012;287:13877–88. [PubMed] DOI:10.1074/jbc.M112.345280
- Hong L, Kim IH, Tombola F. Molecular determinants of Hv1 proton channel inhibition by guanidine derivatives. Proc Natl Acad Sci U S A 2014;111:9971–6. [PubMed] DOI:10.1073/pnas.1324012111
- Yang S, Zhang JJ, Huang XY, et al. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009;15:124–34. [PubMed] DOI:10.1016/j.ccr.2008.12.019
- Dhennin-Duthille I, Gautier M, Faouzi M, et al. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: correlation with pathological parameters. Cell Physiol Biochem 2011;28:813–22. [PubMed] DOI:10.1159/000335795
- Meng X, Cai C, Wu J, et al. TRPM7 mediates breast cancer cell migration and invasion through the MAPK pathway. Cancer Lett 2013;333:96–102. [PubMed] DOI:10.1016/j.canlet.2013.01.031
- Middelbeek J, Kuipers AJ, Henneman L, et al. TRPM7 is required for breast tumor cell metastasis. Cancer Res 2012;72:4250–61. [PubMed] DOI:10.1158/0008-5472.CAN-11-3863
- Guilbert A, Gautier M, Dhennin-Duthille I, et al. Transient receptor potential melastatin 7 is involved in oestrogen receptor-negative metastatic breast cancer cells migration through its kinase domain. Eur J Cancer 2013;49:3694–707. [PubMed] DOI:10.1016/j.ejca.2013.07.008
- Choe S, Kreusch A, Pfaffinger PJ. Towards the three-dimensional structure of voltage-gated potassium channels. Trends Biochem Sci 1999;24:345–9. [PubMed] DOI:10.1016/S0968-0004(99)01440-1
- Potassium channels and chloride channels. In: Hille B. Ion Channels of Excitable Membranes, 3rd edition. Sunderland: Sinauer Associates, 2001:134-41.
- Yellen G. The voltage-gated potassium channels and their relatives. Nature 2002;419:35–42. [PubMed] DOI:10.1038/nature00978
- Huang X, Jan LY. Targeting potassium channels in cancer. J Cell Biol 2014;206:151–62. [PubMed] DOI:10.1083/jcb.201404136
- Bielanska J, Hernández-Losa J, Pérez-Verdaguer M, et al. Voltage-dependent potassium channels Kv1.3 and Kv1.5 in human cancer. Curr Cancer Drug Targets 2009;9:904–14. [PubMed] DOI:10.2174/156800909790192400
- Hammadi M, Chopin V, Matifat F, et al. Human ether à-gogo K+ channel 1 (hEag1) regulates MDA-MB-231 breast cancer cell migration through Orai1-dependent calcium entry . J Cell Physiol 2012;227:3837–46. [PubMed] DOI:10.1002/jcp.24095
- Meng Q, Xia C, Fang J, et al. Role of PI3K and AKT specific isoforms in ovarian cancer cell migration, invasion and proliferation through the p70S6K1 pathway. Cell Signal 2006;18:2262–71. [PubMed] DOI:10.1016/j.cellsig.2006.05.019
- Zhang Z, Li Z, Gao C, et al. miR-21 plays a pivotal role in gastric cancer pathogenesis and progression. Lab Invest 2008;88:1358–66. [PubMed] DOI:10.1038/labinvest.2008.94
- Leung YM, Wong KL, Lin CH, et al. Dependence of 6β-acetoxy-7α- hydroxyroyleanone block of Kv1.2 channels on C-type inactivation. Cell Mol Life Sci 2010;67:147–56. DOI:10.1007/s00018-009-0178-0
- Leung YM, Huang CF, Chao CC, et al. Voltage-gated K+ channels play a role in cAMP-stimulated neuritogenesis in mouse neuroblastoma N2A cells . J Cell Physiol 2011;226:1090–8. [PubMed] DOI:10.1002/jcp.22430
- Aissaoui D, Mlayah-Bellalouna S, Jebali J, et al. Functional role of Kv1.1 and Kv1.3 channels in the neoplastic progression steps of three cancer cell lines, elucidated by scorpion peptides. Int J Biol Macromol 2018;111:1146–55. [PubMed] DOI:10.1016/j.ijbiomac.2018.01.144
- Zeng W, Liu Q, Chen Z, et al. Silencing of hERG1 gene inhibits proliferation and invasion, and induces apoptosis in human osteosarcoma cells by targeting the NF-κB Pathway. J Cancer 2016;7:746–57. DOI:10.7150/jca.13289
- Weaver AK, Bomben VC, Sontheimer H, et al. Expression and function of calcium-activated potassium channels in human glioma cells. Glia 2006;54:223–33. [PubMed] DOI:10.1002/glia.20364
- D’Alessandro G, Catalano M, Sciaccaluga M, et al. KCa3.1 channels are involved in the infiltrative behavior of glioblastoma in vivo . Cell Death Dis 2013;4:e773. [PubMed] DOI:10.1038/cddis.2013.279
- Chantôme A, Potier-Cartereau M, Clarysse L, et al. Pivotal role of the lipid Raft SK3-Orai1 complex in human cancer cell migration and bone metastases. Cancer Res 2013;73:4852–61. [PubMed] DOI:10.1158/0008-5472.CAN-12-4572
- Stroka KM, Jiang H, Chen SH, et al. Water permeation drives tumor cell migration in confined microenvironments. Cell 2014;157:611–23. [PubMed] DOI:10.1016/j.cell.2014.02.052
- Pillozzi S, Brizzi MF, Bernabei PA, et al. VEGFR-1 (FLT-1), β1 integrin, and hERG K+ channel for a macromolecular signaling complex in acute myeloid leukemia: role in cell migration and clinical outcome . Blood 2007;110:1238–50. DOI:10.1182/blood-2006-02-003772
- Yu SP. Regulation and critical role of potassium homeostasis in apoptosis. Prog Neurobiol 2003;70:363–86. [PubMed] DOI:10.1016/S0301-0082(03)00090-X
- Yu SP, Yeh CH, Sensi SL, et al. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science 1997;278:114–7. [PubMed] DOI:10.1126/science.278.5335.114
- Hughes FM Jr, Bortner CD, Purdy GD, et al. Intracellular K+ suppresses the activation of apoptosis in lymphocytes . J Biol Chem 1997;272:30567–76. [PubMed] DOI:10.1074/jbc.272.48.30567
- Hughes FM Jr, Cidlowski JA. Potassium is a critical regulator of apoptotic enzymes in vitro and in vivo . Adv Enzyme Regul 1999;39:157–71. [PubMed] DOI:10.1016/S0065-2571(98)00010-7
- Feinshreiber L, Singer-Lahat D, Ashery U, et al. Voltage-gated potassium channel as a facilitator of exocytosis. Ann N Y Acad Sci 2009;1152:87–92. [PubMed] DOI:10.1111/j.1749-6632.2008.03997.x
- Jang SH, Kang KS, Ryu PD, et al. Kv1.3 voltage-gated K+ channel subunit as a potential diagnostic marker and therapeutic target for breast cancer . BMB Rep 2009;42:535–9. [PubMed] DOI:10.5483/BMBRep.2009.42.8.535