
Tumor pyruvate kinase M2: A promising molecular target of gastrointestinal cancer
Introduction
Gastrointestinal (GI) cancer is one of the most common causes of cancer-related deaths worldwide, and accounts for 40.6% of all reported cases (1). The combined incidence of gastric, esophageal, colorectal and pancreatic cancers accounted for more than 30% of all diagnosed cancers in China (2). The high mortality rate associated with GI cancers is due to the advanced stage of the disease at diagnosis. The current tumor cells markers for pathological examination have low sensitivity, and are limited to detecting post-surgery recurrence or monitoring response to treatment. Several researchers have also questioned the clinical utility of the most commonly used GI tumor markers, such as carcinoembryonic antigen (CEA) and carbohydrate antigen 19-9 (CA19-9) (3-5). Therefore, it is essential to identify novel diagnostic markers for the early detection of tumors in order to reduce mortality rates.
In the 1920s, Warburg et al. observed that unlike normal cells, cancer cells were largely dependent on glycolysis instead of mitochondrial oxidative phosphorylation, independent of oxygen availability (6). This process was subsequently called the Warburg effect or aerobic glycolysis, and is one of the hallmarks of cancer cells (7). This metabolic reprogramming in cancer cells generates adenosine triphosphate (ATP) and allows the cells to survive in anoxic conditions, in addition to providing them with the intermediates and substrates for the biosynthesis of nucleotides, proteins and membrane components that are necessary for proliferating cells (8,9). However, glycolysis produces ATP less efficiently than oxidative phosphorylation, and to compensate for this loss of ATP, cancer cells up-regulate the genes encoding glucose transporters and glycolytic enzymes, leading to increased glucose uptake and metabolism (10). Increased glucose uptake can therefore be used for the clinical detection of tumors using fluorodeoxyglucose positron emission tomography (FPET) (11). Studies show that the glycolytic enzyme pyruvate kinase (PK) plays an important role in metabolic reprogramming of tumor cells and the ensuing aerobic glycolysis (12). PK catalyzes the irreversible conversion of phosphoenolpyruvate (PEP) and adenosine diphosphate (ADP) to pyruvate and ATP, which is the final and one of the three rate-limiting steps of glycolysis (13). Once pyruvate is synthesized, it is either fed into the tri-carboxylic acid (TCA) cycle for oxidative phosphorylation or reduced to lactate under anaerobic conditions (9).
PK has several isoforms in mammals, of which the M2 isozyme or PKM2 is a characteristic metabolic marker of tumor cells (12). Several recent studies show the therapeutic potential of PKM2 in GI cancer. In this review, we have summarized the current knowledge on PKM2 expression and regulation, and its role in the occurrence and development of GI tumors. In addition, we have emphasized the unique role of PKM2 in glucose metabolism and its ability to disrupt the metabolic control of cancer cells, and its potential as a therapeutic target for GI cancer.
Regulation of PKM2 expression and activity
In mammals, the PK family consists of four isoforms: PKL (liver, kidneys and intestine), PKR (red blood cells), and PKM1 and PKM2 (muscle) (Figure 1). PKL and PKR are products of the PKLR (liver/red cell pyruvate kinase) gene (14). PKL has the lowest affinity to PEP among all isoforms, while PKR is inhibited by ATP and activated by fructose-1,6-bisphosphate (FBP) (15). PKM1 and PKM2 are produced by alternative splicing of the primary RNA transcript of PKM gene, and contain sequences encoded by exons 9 and 10, respectively (16). The non-allosteric isoform PKM1 is constitutively active and expressed in terminally differentiated tissues like the muscle and brain, which require a large supply of ATP. In contrast, PKM2 is expressed in tissues with anabolic functions, including normal proliferating cells and cancer cells, and is subject to complex allosteric regulation. PKM2 is overexpressed in most cancer cells, making it a suitable target for cancer therapy.
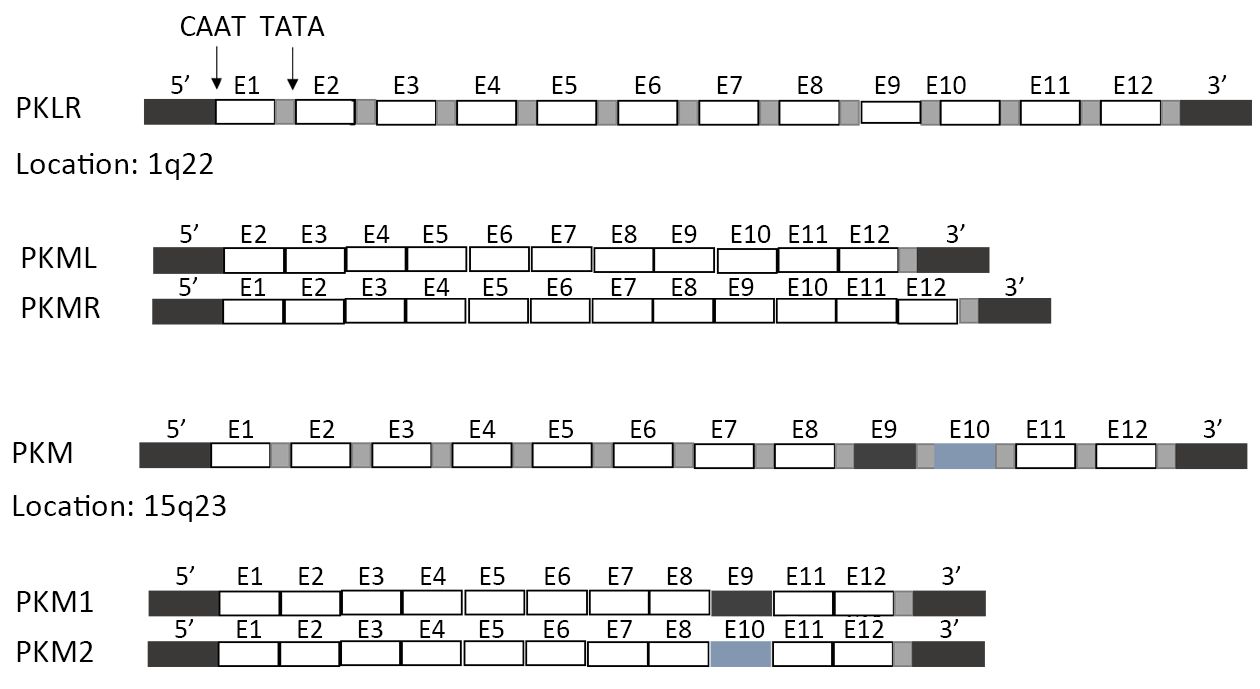
Unlike PKM1 which spontaneously forms a highly active tetramer, PKM2 forms a dimer with very low catalytic efficacy. This low activity explains the preference of the proliferating cells for PKM2, since the latter results in reduced PEP clearance and consequently, a build-up of glycolytic intermediates which are subsequently fluxed into biosynthetic pathways necessary to sustain proliferation. Therefore, the metabolic fate of glucose in highly proliferating tumor cells is largely influenced by the regulation of PKM2.
The expression of PKM2, as well as the ratio between its tetramer and dimer forms, is regulated at various levels through mitogenic signaling. In other words, the cellular metabolic state responds to external signaling cues through changes in PKM2 expression (17). The preferential expression of the M1 and M2 splice variants is controlled by three heterogeneous nuclear ribonucleoproteins, hnRNPI, hnRNPA1 and hnRNPA2, which are transcriptionally regulated by c-Myc (18), which in turn is stabilized by mitogenic signaling cascades like the Ras/Raf/ERK pathway (19). However, c-Myc is often dysregulated in cancer cells via mechanisms that are independent of external signaling cues. For example, studies have documented amplification of the c-Myc gene in cancer cells, which may act as the “driver mutation” and determine the metabolic profile of cancer cells (20). Interestingly, PKM2 has also been shown to translocate to the nucleus where it acts as a cofactor of β-catenin, and induces transcription of c-Myc and subsequently that of key glycolytic proteins such as glucose and lactate transporters (21).
The stoichiometric ratio of the PKM2 monomers, dimers and tetramers is also modulated by post-translational modifications. PKM2 is allosterically activated by the glycolytic intermediate FBP. However, phosphorylation of PKM2 at Tyr105 reduces its binding affinity to FBP, resulting in lower PKM2 activity (22). In addition, direct binding of tyrosine-phosphorylated peptides to PKM2 may also result in decreased FBP binding, and consequently decreased PKM2 tetramerization and activation (23). The PI3k/Akt/mTOR pathway has also been implicated in PKM2 regulation. Treatment of cells with rapamycin (mTOR inhibitor) for example decreased PKM2 expression levels, thereby underscoring the role of external signaling cues in controlling cell metabolism via altering PKM2 activity (24).
The ratio between catalytically active PKM2 tetramers and low-activity dimers is also allosterically controlled by substrate availability (25). In addition to FBP which acts as a cofactor that stabilizes PKM2 tetramers, the latter is also activated by serine (26,27). In rapidly dividing cells, serine levels are low due to its consumption in purine biosynthesis, which dampens PKM2 activation and decreases glucose flux through glycolysis (28). Upregulation of PKM2 in cancer cells with small molecular activators resulted in an increased glycolytic flux with a concomitant drop in glycolytic intermediates and the ability to synthesize serine, thereby rendering cancer cells auxotrophic to serine (29).
In contrast, other metabolites such as phenylalanine and oxalate affect tumor cell growth by inhibiting PKM2 activity (30). A recent study showed that oxalate not only increased the proliferation of breast cancer cells (e.g. the MCF-7 and MDA-MB-231 cell lines), but also enhanced in vivo development of breast tumors in nude mice after direct injection into the mammary fat pad. The tumorigenic effects of oxalate are related its ability to suppress PKM2 activity, which results in the accumulation of metabolic intermediates that are subsequently fluxed into biosynthetic pathways (31).
Functions of PKM2 in GI cancers
PKM2 promotes tumor growth, metastasis and chemo-resistance by regulating either tumor cell metabolism directly as an enzyme or different signaling pathways as a nuclear transcription factor, depending on the tumor type. In the following section, we have highlighted the current research on the role of PKM2 in GI cancers.
PKM2 acts as a proliferative agent
PKM2 promotes the growth of gastric cancer cells by transcriptionally regulating Bcl-xL (32). PKM2 knockdown partially affected the stability of the NF-kB subunit p65, indicating that post-translational regulation of p65 is one of the mechanisms used by PKM2 to drive tumor growth (33). Recent studies have reported presence of PKM2 in the blood of patients with gastrointestinal, pancreatic, lung and ovarian cancer, and renal cell carcinoma (34). Therefore, circulating PKM2 levels can be a potential diagnostic marker for these cancer types. A recent study showed that the dimeric form of PKM2 in blood facilitated tumor growth and neo-angiogenesis by increasing endothelial cell proliferation, migration and adhesion to the extracellular matrix (35).
The pro-proliferative functions of PKM2 depend on its nuclear translocation, which is promoted by different post-translational modifications like tyrosine phosphorylation, lysine acetylation, or sumoylation in response to epidermal growth factor receptor (EGFR), interleukin-3 (IL-3) and Oct-4, respectively (36). In addition to stimulating cell proliferation, PKM2 translocation also promotes apoptosis via a caspase and Bcl-2 independent manner in response to stimuli like DNA damage or oxidative stress (37,38). Furthermore, PKM2 knockout in Eca109 and EC9706 cells activated caspase 3, down-regulated caspase 9, and increased expression of Bim (39).
PKM2 facilitates tumor metastasis
PKM2 is known to increase colorectal cancer (CRC) metastasis by modulating the STAT3 signaling pathway (40). The study of Atsushi Hamabe et al. showed that PKM2 mediated epithelial-mesenchymal transition (EMT), which is critical for cancer cells to acquire the invasive potential. They also demonstrated that EMT stimulated the nuclear translocation of PKM2 and transcriptionally downregulated epithelial cadherin (a requirement for EMT induction). PKM2 also repressed E-cadherin epigenetically by interacting with the transcriptional factor TGF-β-induced factor homeobox 2, which induces histone H3 deacetylation and downregulates E-cadherin transcription. These findings indicate a novel regulatory axis involving PKM2 that can be utilized to prevent cancer metastasis (35).
PKM2 enhances chemo-resistance
Studies have linked dysregulated glucose metabolism with chemo-resistance, and since PKM2 is an important regulator of tumor glycolysis, it has been implicated in drug resistance in different GI cancers. 5-Fluorouracil (5-FU) is an important agent used for the systemic treatment of GI cancers, but has limited clinical response due to emergence of resistance among the patients (41). Studies have linked PKM2 expression and activity to cisplatin resistance in gastric tumor cells and to acquired resistance to 5-FU in CRC cells, indicating its role in acquired chemo-resistance. In addition, PKM2 is a direct target of miR-122 in the colon cancer cells, and overexpression of the latter in 5-FU-resistant cells re-sensitizes them to 5-FU by inhibiting PKM2 (42).
The nucleoside analog gemcitabine (2’,2’-difluorodeoxycytidine) is routinely used in the treatment of advanced pancreatic cancers (43). Kim et al., Calabretta et al. and Pandita et al. have elucidated different mechanisms by which PKM2 can induce gemcitabine chemo-resistance, and have suggested the possibility of combining gemcitabine and a PKM2 inhibitor to improve therapeutic response of pancreatic cancer cells (43-45). Nuclear translocation of PKM2 increases gefitinib resistance in CRC cells via STAT3 activation, and CRC patients who are resistant to gefitinib or oxaliplatin have higher PKM2 expression (40,46). In addition, knockdown of both PKM2 and GLS1 significantly reversed oxaliplatin-resistance in CRC cells (47). PKM2 also modulates the response of esophageal squamous cell carcinoma (ESCC) cells to chemotherapy by regulating the pentose phosphate pathway (48).
PKM2 as a therapeutic target in GI cancers
In view of the important role of PKM2 in GI cancers, it is an attractive therapeutic target in these cancers. Due to the metabolic diversity of cancer cells, we have discussed the prognostic and therapeutic relevance of PKM2 in different GI cancers.
Esophageal cancer
Esophageal cancer is the sixth leading cause of cancer-related deaths and the eighth most common cancer worldwide (49). Surgery alone or in combination with neoadjuvant chemo-radiotherapy, adjuvant radiotherapy, and/or adjuvant chemotherapy is the main therapeutic modality of esophageal cancer (50). A meta-analysis showed that high expression of PKM2 was associated with poor prognosis in esophageal squamous carcinoma, and highlighted its prognostic significance (51). In addition, high PKM2 expression was associated with the clinical stage, tumor stage, nodal metastasis and differentiation, indicating its potential as a prognostic biomarker in esophageal cancer (52). A recent study established PKM2 as a marker of transformed and highly proliferating cells during the metaplasia-dysplasia-adenocarcinoma sequence in Barrett’s esophagus (53).
In the last two decades, tanshione IIA has gained attention for its potential therapeutic effects in cancer. A research investigated the anticancer activity of tanshione IIA in human esophagus cancer Ec109 cells through the inhibition of PKM2 expression (54).
Gastric cancer
Gastric cancer is the fifth most common malignancy diagnosed worldwide, after cancers of the lung, breast, colorectum and prostate (55). PKM2 overexpression in gastric cancer was significantly correlated with certain parameters of tumor progression, such as clinical stage, nodal metastasis, tumor stage and tumor size. However, the high levels of PKM2 were not associated with tumor cell differentiation (34). Recent evidence suggests that anti-diabetic drug metformin prevents cancer progression. Metformin has profound antitumor effect by inducing intrinsic apoptosis via the inhibition of HIF1α/PKM2 signaling pathway in gastric cancer cell SGC7901 and BGC823 (56).
Pancreatic cancer
Pancreatic cancer is the fourth leading cause of cancer-related deaths world-wide. Only 10%−15% of pancreatic cancer patients have survival chances by surgical resection. Therefore, it is an urgent research to find therapeutic targets for pancreatic cancer.
Chronic exposure to gemcitabine leads to modulation of PKM alternative splicing in pancreatic ductal adenocarcinoma cells, conferring resistance to the drug. It is highly feasible to combine gemcitabine and a PKM2 inhibitor in order to improve chemotherapeutic response in pancreatic cancer (45,57).
CRC
CRC is the third most common cancer in males and the second-most prevalent in females (58). High levels of fecal PKM2 have been proposed as a novel diagnostic biomarker for detecting CRC, with a sensitivity of 64% in detecting early stage (T1 and T2) cancer. However, a recent systematic review and meta-analysis showed ambiguous sensitivity and specificity of this marker (59).
As mentioned above, nuclear PKM2 modulated the sensitivity of CRC cells to gefitinib and indicated that small molecule pharmacological disruption of nuclear PKM2 association with STAT3 is a potential avenue for overcoming EGFR-TKI resistance in CRC patients (40,46).
Conclusions
Cancer is caused by the uncontrolled proliferation of cells, which substantially increases the demand for nutrients and cellular building blocks, and therefore requires metabolic changes. Targeting the unique metabolic characteristics of cancer cells, such as absorption of amino acids, glutamine and aerobic glycolysis, has therefore emerged as a novel selective therapeutic strategy against cancer cells. Under physiological conditions, glucose is decomposed into pyruvic acid via glycolysis, which then is fed into the mitochondrial TCA cycle and completely oxidized to carbon dioxide. Cancer cells however metabolize most of the glucose anaerobically, independent of oxygen availability, and the resulting pyruvate is converted into lactic acid and secreted from the cells. It is not completely clear as to why cancer cells use this inefficient method of producing energy, but it is hypothesized that this metabolic pathway helps the cancer cells use nutrients more effectively for proliferation.
PKM2 catalyzes one of the rate-limiting steps of glycolysis, and its overexpression in tumor cells is associated with glycolysis, tumorigenesis, metastasis and chemo-resistance. In addition, high levels of PKM2 in solid tumors are associated with poor prognosis. A strong correlation has been reported between plasma/fecal PKM2 levels and the tumor stage in gastric, colorectal and pancreatic cancers, indicating its potential as a prognostic biomarker and therapeutic target in GI cancers. There should be no illusions, targeting cancer metabolism by PKM2 represents novel opportunities for enhancing therapeutic efficacy. However, PKM2 as a therapeutic target for clinical treatment still needs more experimental verification.
Acknowledgements
This work was supported by the grants from ‘San Ming’ Project of Shenzhen city, China (No. SZSM201612051); Municipal Health Planning Commission Fund of Shenzhen city, China (No. 201601004, No. SZXJ2017078 and No. SXZJ2018084).
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [PubMed] DOI:10.3322/caac.21492
- Chen W, Zheng R, Zuo T, et al. National cancer incidence and mortality in China, 2012. Chin J Cancer Res 2016;28:1–11. [PubMed] DOI:10.3978/j.issn.1000-9604.2016.02.08
- Pokorny RM, Hunt L, Galandiuk S. What’s new with tumor markers for colorectal cancer?. Dig Surg 2000;17:209–15. [PubMed] DOI:10.1159/000018853
- McLaughlin R, O’Hanlon D, Kerin M, et al. Are elevated levels of the tumour marker CA19-9 of any clinical significance? — an evaluation. Ir J Med Sci 1999;168:124–6. [PubMed] DOI:10.1007/BF02946481
- Mann DV, Edwards R, Ho S, et al. Elevated tumour marker CA19-9: clinical interpretation and influence of obstructive jaundice. Eur J Surg Oncol 2000;26:474–9. [PubMed] DOI:10.1053/ejso.1999.0925
- Warburg O. On the origin of cancer cells. Science 1956;123:309–14. [PubMed] DOI:10.1126/science.123.3191.309
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [PubMed] DOI:10.1016/j.cell.2011.02.013
- Mathupala SP, Rempel A, Pedersen PL. Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J Biol Chem 2001;276:43407–12. [PubMed] DOI:10.1074/jbc.M108181200
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324:1029–33. DOI:10.1126/science.1160809
- Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell 2008;134:703–7. [PubMed] DOI:10.1016/j.cell.2008.08.021
- Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nat Rev Cancer 2002;2:683–93. [PubMed] DOI:10.1038/nrc882
- Chaneton B, Gottlieb E. Rocking cell metabolism: revised functions of the key glycolytic regulator PKM2 in cancer. Trends Biochem Sci 2012;37:309–16. [PubMed] DOI:10.1016/j.tibs.2012.04.003
- Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol 2011;43:969–80. [PubMed] DOI:10.1016/j.biocel.2010.02.005
- Harada K, Saheki S, Wada K, et al. Purification of four pyruvate kinase isozymes of rats by affinity elution chromatography. Biochim Biophys Acta 1978;524:327–39. [PubMed] DOI:10.1016/0005-2744(78)90169-9
- Yamada K, Noguchi T. Nutrient and hormonal regulation of pyruvate kinase gene expression. Biochem J 1999;337:1–11. [PubMed] DOI:10.1042/bj3370001
- Noguchi T, Inoue H, Tanaka T. The M1- and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J Biol Chem 1986;261:13807–12. [PubMed]
- Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab 2012;23:560–6. [PubMed] DOI:10.1016/j.tem.2012.06.010
- David CJ, Chen M, Assanah M, et al. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010;463:364–8. [PubMed] DOI:10.1038/nature08697
- Sears R, Nuckolls F, Haura E, et al. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 2000;14:2501–14. [PubMed] DOI:10.1101/gad.836800
- Little CD, Nau MM, Carney DN, et al. Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature 1983;306:194–6. [PubMed] DOI:10.1038/306194a0
- Yang W, Zheng Y, Xia Y, et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol 2012;14:1295–304. DOI:10.1038/ncb2629
- Hitosugi T, Kang S, Vander Heiden MG, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal 2009;2:ra73. [PubMed] DOI:10.1126/scisignal.2000431
- Christofk HR, Vander Heiden MG, Wu N, et al. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008;452:181–6. [PubMed] DOI:10.1038/nature06667
- Nemazanyy I, Espeillac C, Pende M, et al. Role of PI3K, mTOR and Akt2 signalling in hepatic tumorigenesis via the control of PKM2 expression. Biochem Soc Trans 2013;41:917–22. [PubMed] DOI:10.1042/BST20130034
- Wang P, Sun C, Zhu T, et al. Structural insight into mechanisms for dynamic regulation of PKM2. Protein Cell 2015;6:275–87. [PubMed] DOI:10.1007/s13238-015-0132-x
- Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry 2005;44:9417–29. [PubMed] DOI:10.1021/bi0474923
- Chaneton B, Hillmann P, Zheng L, et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 2012;491:458–62. [PubMed] DOI:10.1038/nature11540
- Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer 2016;16:650–62. DOI:10.1038/nrc.2016.81
- Kung C, Hixon J, Choe S, et al. Small molecule activation of PKM2 in cancer cells induces serine auxotrophy. Chem Biol 2012;19:1187–98. [PubMed] DOI:10.1016/j.chembiol.2012.07.021
- Morgan HP, O’Reilly FJ, Wear MA, et al. M2 pyruvate kinase provides a mechanism for nutrient sensing and regulation of cell proliferation. Proc Natl Acad Sci U S A 2013;110:5881–6. DOI:10.1073/pnas.1217157110
- Castellaro AM, Tonda A, Cejas HH, et al. Oxalate induces breast cancer. BMC Cancer 2015;15:761. [PubMed] DOI:10.1186/s12885-015-1747-2
- Kwon OH, Kang TW, Kim JH, et al. Pyruvate kinase M2 promotes the growth of gastric cancer cells via regulation of Bcl-xL expression at transcriptional level. Biochem Biophys Res Commun 2012;423:38–44. [PubMed] DOI:10.1016/j.bbrc.2012.05.063
- Azoitei N, Becher A, Steinestel K, et al. PKM2 promotes tumor angiogenesis by regulating HIF-1α through NF-κB activation. Mol Cancer 2016;15:3. [PubMed] DOI:10.1186/s12943-015-0490-2
- Wu J, Hu L, Chen M, et al. Pyruvate kinase M2 overexpression and poor prognosis in solid tumors of digestive system: evidence from 16 cohort studies. Onco Targets Ther 2016;9:4277–88. [PubMed] DOI:10.2147/ott.s106508
- Hamabe A, Konno M, Tanuma N, et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc Natl Acad Sci U S A 2014;111:15526–31. [PubMed] DOI:10.1073/pnas.1407717111
- Chen L, Shi Y, Liu S, et al. PKM2: the thread linking energy metabolism reprogramming with epigenetics in cancer. Int J Mol Sci 2014;15:11435–45. [PubMed] DOI:10.3390/ijms150711435
- Feng J, Ma T, Ge Z, et al. PKM2 gene regulates the behavior of pancreatic cancer cells via mitogen-activated protein kinase pathways. Mol Med Rep 2015;11:2111–7. [PubMed] DOI:10.3892/mmr.2014.2990
- Liang J, Cao R, Wang X, et al. Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Res 2017;27:329–51. [PubMed] DOI:10.1038/cr.2016.159
- Tang JC, An R, Jiang YQ, et al. Effects and mechanisms of metformin on the proliferation of esophageal cancer cells in vitro and in vivo . Cancer Res Treat 2017;49:778–89. [PubMed] DOI:10.4143/crt.2015.485
- Li Q, Zhang D, Chen X, et al. Nuclear PKM2 contributes to gefitinib resistance via upregulation of STAT3 activation in colorectal cancer. Sci Rep 2015;5:16082. [PubMed] DOI:10.1038/srep16082
- Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 2003;3:330–8. [PubMed] DOI:10.1038/nrc1074
- He J, Xie G, Tong J, et al. Overexpression of MicroRNA-122 re-sensitizes 5-FU-resistant colon cancer cells to 5-FU through the inhibition of PKM2 in vitro and in vivo . Cell Biochem Biophys 2014;70:1343–50. DOI:10.1007/s12013-014-0062-x
- Kim DJ, Park YS, Kang MG, et al. Pyruvate kinase isoenzyme M2 is a therapeutic target of gemcitabine-resistant pancreatic cancer cells. Exp Cell Res 2015;336:119–29. [PubMed] DOI:10.1016/j.yexcr.2015.05.017
- Calabretta S, Bielli P, Passacantilli I, et al. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene 2016;35:2031–9. [PubMed] DOI:10.1038/onc.2015.270
- Pandita A, Kumar B, Manvati S, et al. Synergistic combination of gemcitabine and dietary molecule induces apoptosis in pancreatic cancer cells and down regulates PKM2 expression. PLoS One 2014;9:e107154. [PubMed] DOI:10.1371/journal.pone.0107154
- Ginés A, Bystrup S, Ruiz de Porras V, et al. PKM2 subcellular localization is involved in oxaliplatin resistance acquisition in HT29 human colorectal cancer cell lines. PLoS One 2015;10:e0123830. [PubMed] DOI:10.1371/journal.pone.0123830
- Lu WQ, Hu YY, Lin XP, et al. Knockdown of PKM2 and GLS1 expression can significantly reverse oxaliplatin-resistance in colorectal cancer cells. Oncotarget 2017;8:44171–85. [PubMed] DOI:10.18632/oncotarget.17396
- Fukuda S, Miyata H, Miyazaki Y, et al. Pyruvate kinase M2 modulates esophageal squamous cell carcinoma chemotherapy response by regulating the pentose phosphate pathway. Ann Surg Oncol 2015;22(Suppl 3):S1461–8. [PubMed] DOI:10.1245/s10434-015-4522-3
- Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87–108. DOI:10.3322/caac.21262
- Pennathur A, Gibson MK, Jobe BA, et al. Oesophageal carcinoma. Lancet 2013;381:400–12. [PubMed] DOI:10.1016/S0140-6736(12)60643-6
- Zhu H, Luo H, Zhu X, et al. Pyruvate kinase M2 (PKM2) expression correlates with prognosis in solid cancers: a meta-analysis. Oncotarget 2017;8:1628–40. [PubMed] DOI:10.18632/oncotarget.13703
- Li W, Xu Z, Hong J, et al. Expression patterns of three regulation enzymes in glycolysis in esophageal squamous cell carcinoma: association with survival. Med Oncol 2014;31:118. DOI:10.1007/s12032-014-0118-1
- Zhang X, He C, He C, et al. Nuclear PKM2 expression predicts poor prognosis in patients with esophageal squamous cell carcinoma. Pathol Res Pract 2013;209:510–5. [PubMed] DOI:10.1016/j.prp.2013.06.005
- Zhang HS, Zhang FJ, Li H, et al. Tanshinone ⅡA inhibits human esophageal cancer cell growth through miR-122-mediated PKM2 down-regulation. Arch Biochem Biophys 2016;598:50–6. [PubMed] DOI:10.1016/j.abb.2016.03.031
- Yang L, Zheng R, Wang N, et al. Incidence and mortality of stomach cancer in China, 2014. Chin J Cancer Res 2018;30:291–8. [PubMed] DOI:10.21147/j.issn.1000-9604.2018.03.01
- Chen G, Feng W, Zhang S, et al. Metformin inhibits gastric cancer via the inhibition of HIF1α/PKM2 signaling. Am J Cancer Res 2015;5:1423–34. [PubMed]
- Tian S, Li P, Sheng S, et al. Upregulation of pyruvate kinase M2 expression by fatty acid synthase contributes to gemcitabine resistance in pancreatic cancer. Oncol Lett 2018;15:2211–7. [PubMed] DOI:10.3892/ol.2017.7598
- Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin 2010;60:277–300. [PubMed] DOI:10.3322/caac.20073
- Li R, Liu J, Xue H, et al. Diagnostic value of fecal tumor M2-pyruvate kinase for CRC screening: a systematic review and meta-analysis. Int J Cancer 2012;131:1837–45. [PubMed] DOI:10.1002/ijc.27442