
Serum LDH level may predict outcome of chronic lymphocytic leukemia patients with a 17p deletion: a retrospective analysis of prognostic factors in China
Introduction
Chronic lymphocytic leukemia (CLL), with an age-adjusted incidence of 4.8 new cases per 100,000 inhabitants per year in the US (1) and an incidence of 0.2–0.3 new cases per 100,000 inhabitants per year in Asia (2), is characterized by a heterogeneous clinical course.
Cytogenetic abnormalities are a major determinant of outcome in patients with CLL (3), among which a 17p deletion (17p-) is considered to have a consistently poor impact on the patients’ prognosis, with an incidence of 4%–7% of newly diagnosed CLL cases and a median survival of 2–3 years from the initial diagnosis (4-6). It was suggested that a 17p- CLL may be particularly resistant to treatment with purine analogues (5) or rituximab (7), which indicated that patients with 17p- CLL should be treated differently from other patients with CLL (8), with an emphasis on therapy using alternative agents, such as alemtuzumab (9) or high-dose methylprednisolone (HDMP) (10).
In addition, the outcome of patients with a 17p- is not always dismal, and a proportion of them may remain asymptomatic for a prolonged period of time (11), which indicates the presence of heterogeneity within the subgroup of patients with a 17p-.
Due to the low incidence, large studies of CLL patients with a 17p- are lacking, especially in the Asian population. Therefore, in the present study, taking advantage of a cohort of single-institutional cases, we attempted to describe the characteristics of patients with a 17p- CLL and the outcome of these patients through different therapeutic approaches and further identify the prognostic factors within this subgroup.
Materials and methods
Patients
A total of 456 treatment-naive CLL patients, who had been diagnosed in the Institute of Hematology and Blood Disease Hospital between March 2000 and January 2015, were included in this study. The baseline characteristics, date of initial therapy, treatment response, and long-term outcome of these patients were confirmed by a manual chart review and follow-up telephone calls. The diagnosis in each case was confirmed according to the World Health Organization (WHO) classification (12). Evidence of persistent lymphocytosis and a compatible immunophenotype were required for the diagnosis. In all the cases, an immunophenotypic analysis was performed by flow cytometry, including at least the following markers: CD19, CD5, CD22, CD23, CD38, CD25, CD103, CD11c, FMC7, BCL2, CD10, and CD20 as well as surface immunoglobulin κ and λ. All patients provided informed consent in accordance with the requirements of the Declaration of Helsinki, and the research project was approved by the Institutional Ethics Review Board of Institute of Hematology and Blood Disease Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College. For this study, the clinical follow-up data were available until the beginning of February 2015. The therapeutic indications were standardized according to the International Workshop on Chronic Lymphocytic Leukemia (IWCLL) criteria (13). The therapeutic regimens during the course of their disease were heterogeneous at the discretion of the treating physician, including: 1) chlorambucil (0.4 mg/kg, d 1–2, d 15–16, or 0.1–0.2 mg/kg, d 1–7 every 28 d, for a maximum of six courses); 2) fludarabine 25 mg/m2 intravenously and cyclophosphamide 250 mg/m2 intravenously on d 1–3 (FC) or in combination with rituximab at 375 mg/m2 on d 0 (FCR); both regimens were repeated every 28 d for a maximum of six courses; 3) cyclophosphamide 750 mg/m2 intravenously on d 1, vincristine 1.4 mg/m2 intravenously on d 1 and prednisone 100 mg/d orally d 1–5 (CVP) plus doxorubicin 50 mg/m2 intravenously on d 1 (CHOP) or in combination with rituximab at 375 mg/m2 on d 0 (R-CVP/R-CHOP); both regimens were repeated every 21 d for a maximum of six courses; 4) methylprednisolone 1 g/m2 for 5 d in combination with rituximab on d 1 at 375 mg/m2 weekly for four 4-week cycles (R-HDMP) or chemotherapy combined with alemtuzumab (30 mg intravenously on d 1, 3 and 5); and 5) other regimens.
Fluorescence in situ hybridization (FISH)
FISH analysis with specific probes for conventional cytogenetic abnormalities was performed. The CLL FISH ‘panel’ included probes for 12 centromere (CEP12), 13q14.3 (LSI D13S25 and RB-1), 14q32 (LSI IGHC/IGHV), 17p13 (LSI TP53), and 11q22 (LSI ATM). The sample preparations and hybridizations were conducted following the manufacturer’s recommendations and as previously described (14,15). The LSI CCND1/IGH Dual Color, Dual Fusion Translocation Probe was used to exclude the possibility of mantle cell lymphoma in the case of t (14q32) positive. All probes were purchased from Vysis (Abbott Co., Downers Grove, IL, USA). The signal screening was carried out on at least 200 cells with well delineated signals. The cut-off values (mean+3×SD) determined from samples of 20 cytogenetically normal people were as follows: 7.5% for CEP 12 and 6.5% for the deletion of D13S25, RB1, and ATM; 6.8% for 17p- and 4.5% for IgH translocation and CCND1/IGH.
Survival and statistical analysis
Time to first therapy (TTFT) was defined as the time between the date of diagnosis and the date of the initiation of the first treatment, death, or the last follow-up for patients untreated. Progression-free survival (PFS) was defined as the interval between the date of diagnosis and the date of disease progression or death from any cause. Time to progression (TTP) for patients responding to treatment was calculated from the date of therapy to the date of disease progression or death. Overall survival (OS) was measured from the date of diagnosis to the date of death or the last follow-up (censoring). Baseline characteristics and prognostic factors were compared using Chi-square or Fisher’s exact tests for discrete variables, and the Mann-Whitney test was used for continuous variables. Kaplan-Meier methodology and a Log-rank test or Cox’s regression were undertaken for survival analyses using the IBM SPSS Statistics (Version 19.0; IBM Corp., New York, USA). Variables that were significant on univariate analysis were further included on multivariate analysis with Cox’s regression. All tests were two-sided. An effect was considered statistically significant if P<0.05.
Receiver-operator characteristic (ROC) analysis and Youden index were applied to determine the optimal cut-off point for 17p- by FISH (15). The cut-off value that best discriminated between survival and death was used for OS, and the cut-off value that best discriminated between progression and no progression was used for PFS.
Results
Clinical characteristics of patients
The clinical characteristics of the study population are shown in Table 1. A total of 456 patients with CLL were analyzed by FISH to detect 17p- and followed up after the diagnosis. We identified 66 (14.5%) patients with de novo 17p-. The median age was 54 years in the 17p- subgroup with a male dominance. Compared to patients without this aberration, patients with 17p- had a more advanced disease at diagnosis, as suggested by an advanced Binet stage, lower hemoglobin level, higher lactic dehydrogenase (LDH) level (≥230 IU/L), higher proportion of B symptoms and a trend towards unmutated immunoglobulin heavy-chain variable region (IGHV) gene status (Table 1).
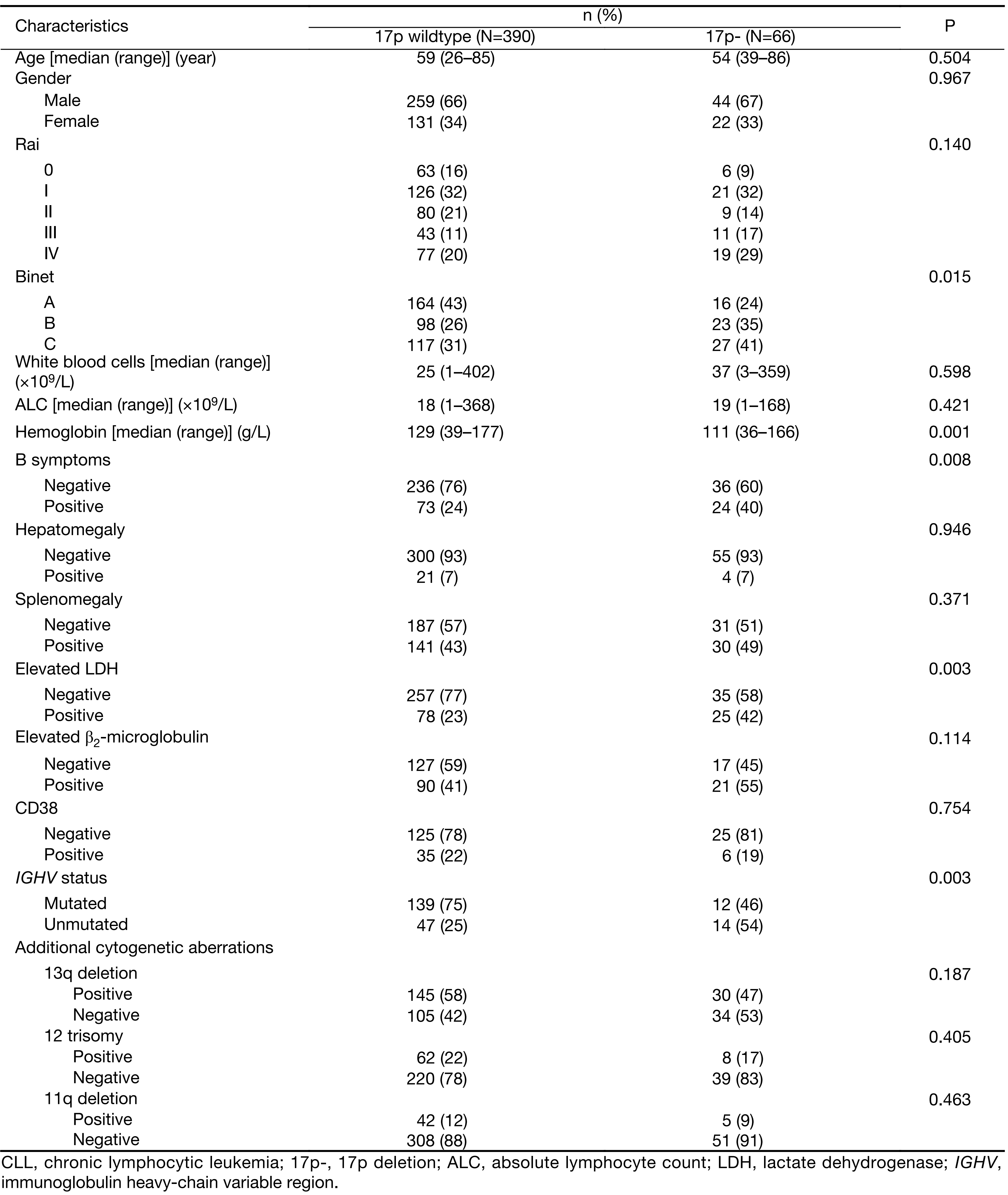
Full table
Therapy
There were no protocols specific for the first-line treatment of patients with 17p- during the study period, and therapy was initiated at a median of 12.0 (range, 0.3–81.0) months after the diagnosis of CLL. Table 2 shows the response rate for the first-line therapy in 51 patients with documented treatment outcomes, and the response rate was stratified according to the clonal size of the 17p-. The most frequently used therapies were chlorambucil (25.5%), FC (25.5%), FCR (15.7%), rituximab + chemotherapy not including fludarabine (9.8%), and chemotherapy not including fludarabine (9.8%). The overall response rate (ORR) and complete response rate (CRR) in patients with 17p- were 56.9% and 17.0%, respectively, which were lower than those in patients with negative 17p- (ORR 67.9% and CRR 24.4%), but no significant difference was observed between them. Furthermore, patients with a 17p- were divided into two subgroups according to the percentage of leukemic cells carrying a 17p- as follows: a high percentage 17p- subgroup (defined as having ≥25% of cells with a 17p-) and a low percentage 17p- subgroup (defined as having <25% of cells harbouring a 17p-). There was a tendency towards an even lower response rate in patients with a high percentage of 17p-, and the TTP was significantly shorter in patients with a high percentage of 17p- compared to a low percentage 17p- (8.5 months vs. 68.0 months, P<0.001).
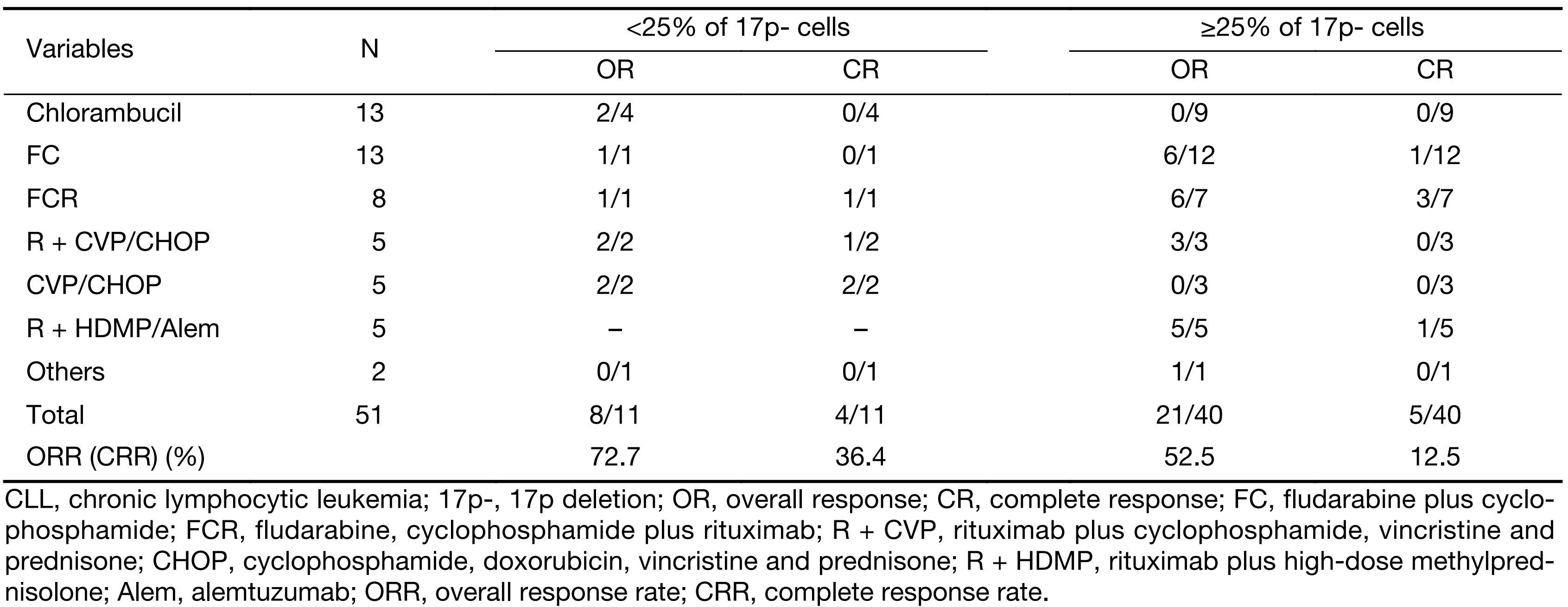
Full table
Patients with a partial response (PR) had a short TTP of only 15 months, which was not significantly better than that of non-responders, whereas patients with complete response (CR) had a significantly superior TTP (55.6% progression free at 4 years, P=0.023 for all comparisons, see Figure 1A). The superiority in disease control translated to an improved survival after the initiation of therapy (Figure 1B) as follows: patients achieving CR had favorable survival, whereas patients with no response to initial therapy had a significantly inferior outcome (median 96.0 and 26.0 months, respectively, P=0.002). For patients with PR, the median survival was 78.0 months, and the improvement was not significant compared to patients who failed to respond to the initial therapy (P=0.086).
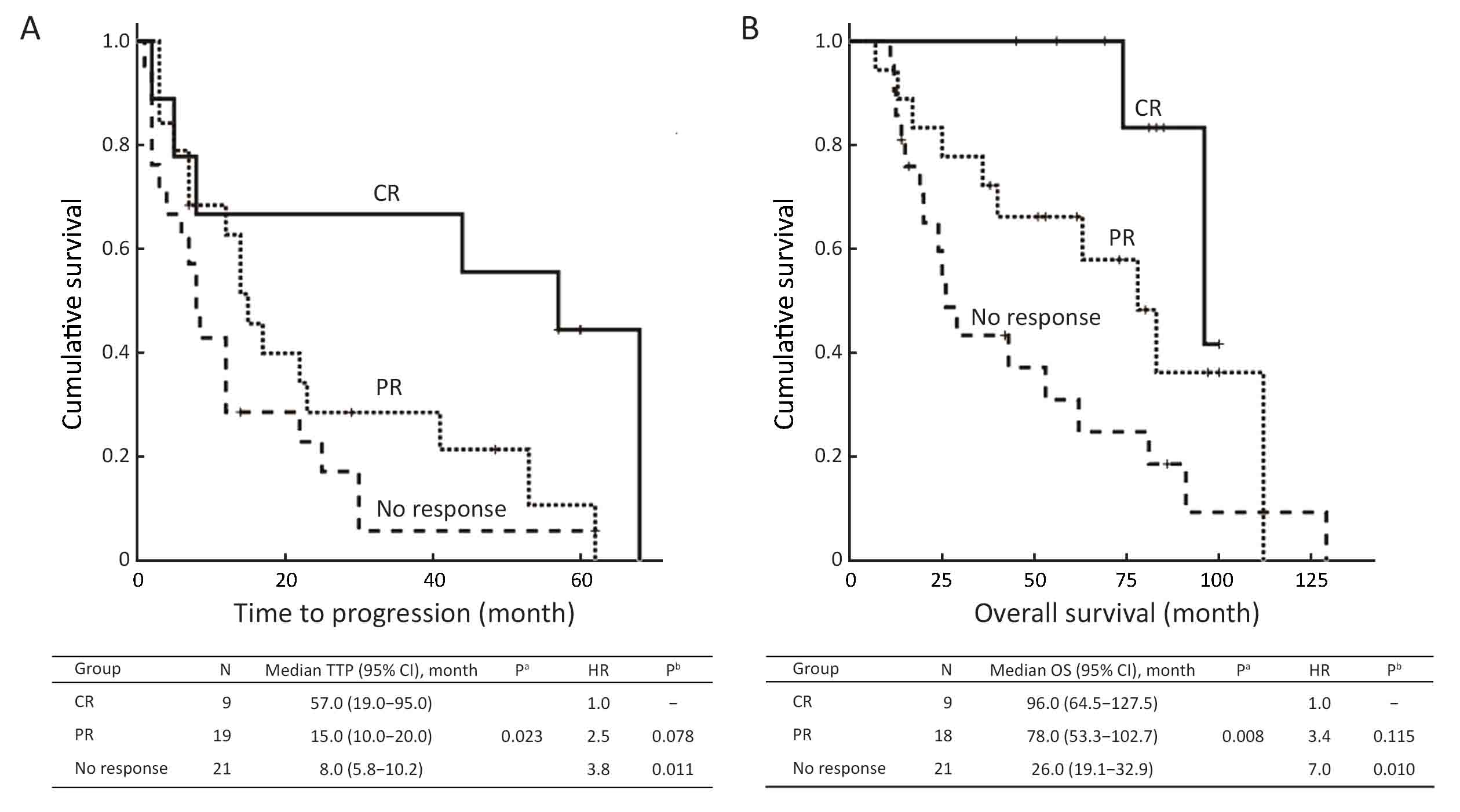
Prognosis according to percentage of nuclei with a 17p-
The median follow-up time is 45.0 (range, 2.0–288.0) months for the cohort of patients. Of the 66 patients with a 17p-, 6 patients (9.1%) were lost to follow-up, 56 (84.8%) received chemotherapy, 44 (66.7%) had disease progression and 32 (48.5%) died. Of the 390 patients testing negative for a 17p-, 15 patients (3.8%) were lost to follow-up, 240 (61.5%) received chemotherapy, 118 (30.3%) had disease progression and 73 (18.7%) died.
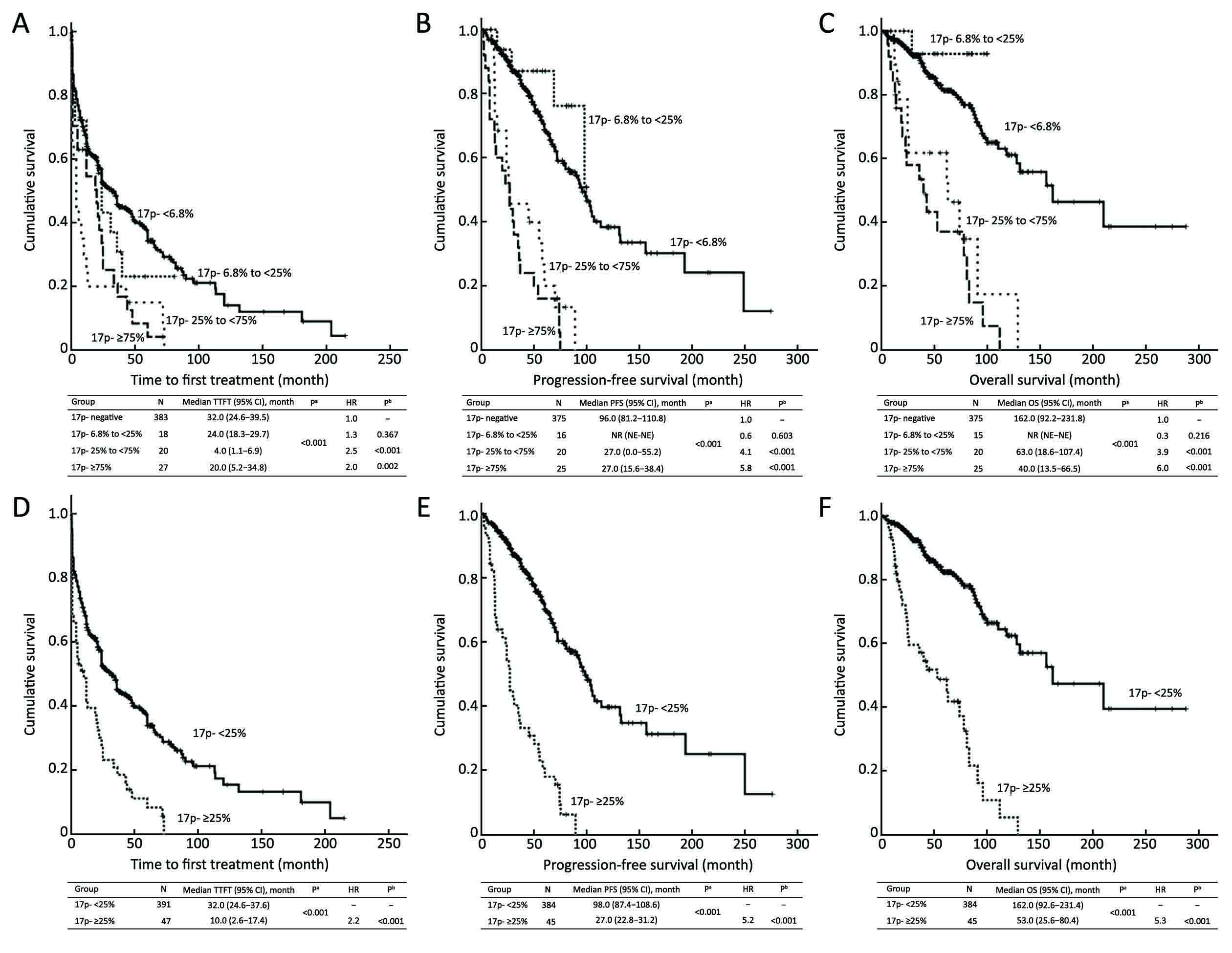
The estimated median TTFT for patients positive for a 17p- was 21.0 [95% confidence interval (95% CI): 9.7–32.3] months, which is significantly shorter than that for patients without a 17p- (49.0 months, 95% CI: 39.0–59.0 months; P<0.001). There was a significant difference between the two groups regarding PFS (median PFS, 96.0 months in 17p- negative group vs. 36.0 months in 17p- positive one; P<0.001). In contrast, the 5-year OS was significantly higher in the 17p- negative group than in the 17p- positive group (81.4% vs. 57.4%; median OS, 162.0 vs. 78.0 months; P<0.001).
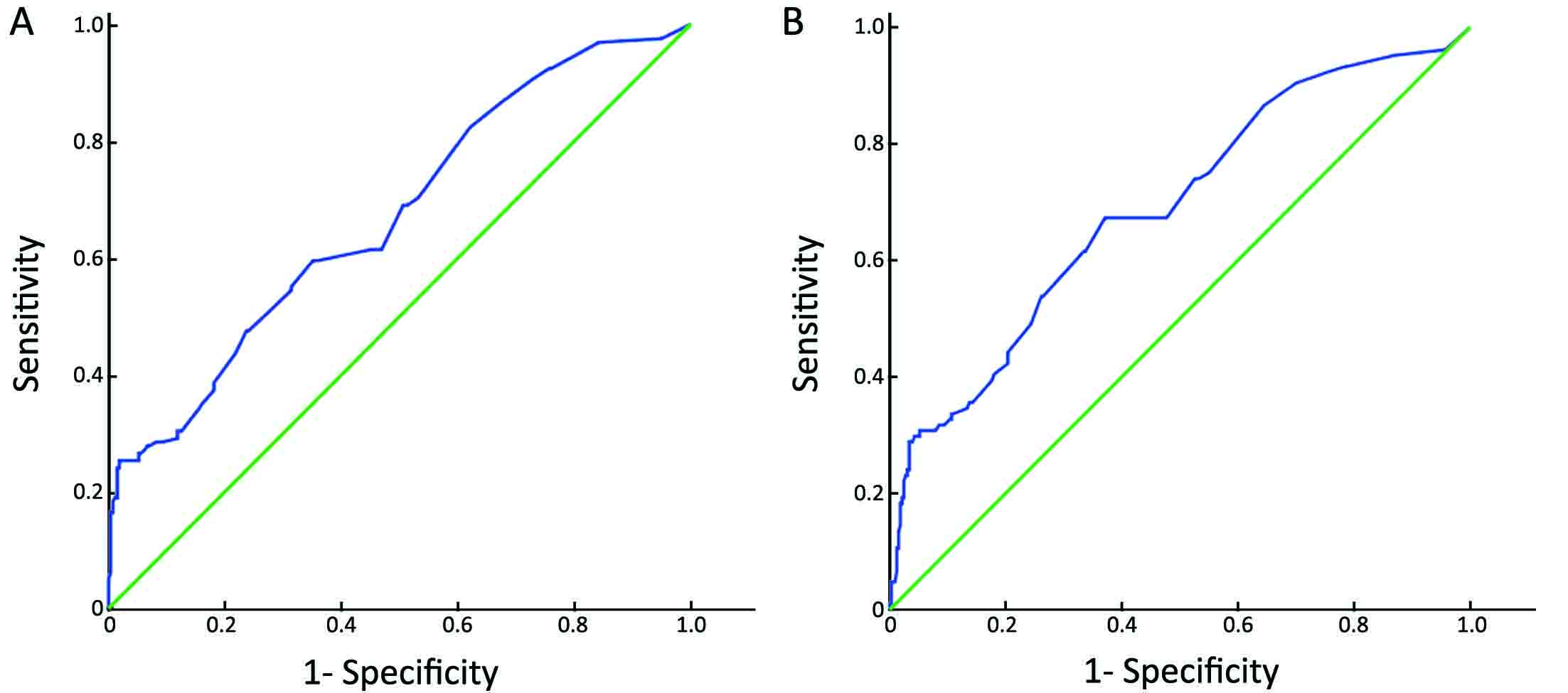
In the current study, the median percentage of CLL cells harboring a 17p- was 2.0% (range, 0–99.8%). The survival curves of TTFT, PFS and OS, according to the clonal size of the 17p-, are shown in Figure 2A, B, C. Of note, the percentage of involved cells at almost any cut-off point was tested. However, the ROC analysis revealed that the cut-off point of 24.25% provided the highest Youden values with regard to PFS and OS (Supplementary Figure S1). Meanwhile, patients with 6.8%–25.0% involved cells had a similar survival as patients harboring a negative 17p- (P=0.303 as regards PFS and P=0.181 as regards OS). Therefore, we used 25% to divide the patients into the two subgroups. The median TTFT of patients with <25% and ≥25% 17p- cells was 32.0 (95% CI: 26.4–37.6) months and 10.0 (95% CI: 2.6–17.4) months, respectively (P<0.001), and the median PFS for these two subgroups was 98.0 (95% CI: 87.4–108.6) months and 27.0 (95% CI: 22.8–31.2) months, respectively (P<0.001). In addition, the median OS of patients with <25% and ≥25% 17p- cells was 162.0 (95% CI: 92.6–231.4) months and 53.0 (95% CI: 25.6–80.4) months, respectively (P<0.001). The survival curves using 25% as a cut-off value are shown in Figure 2D, E, F.
Univariate and multivariate analyses within 17p- subgroup
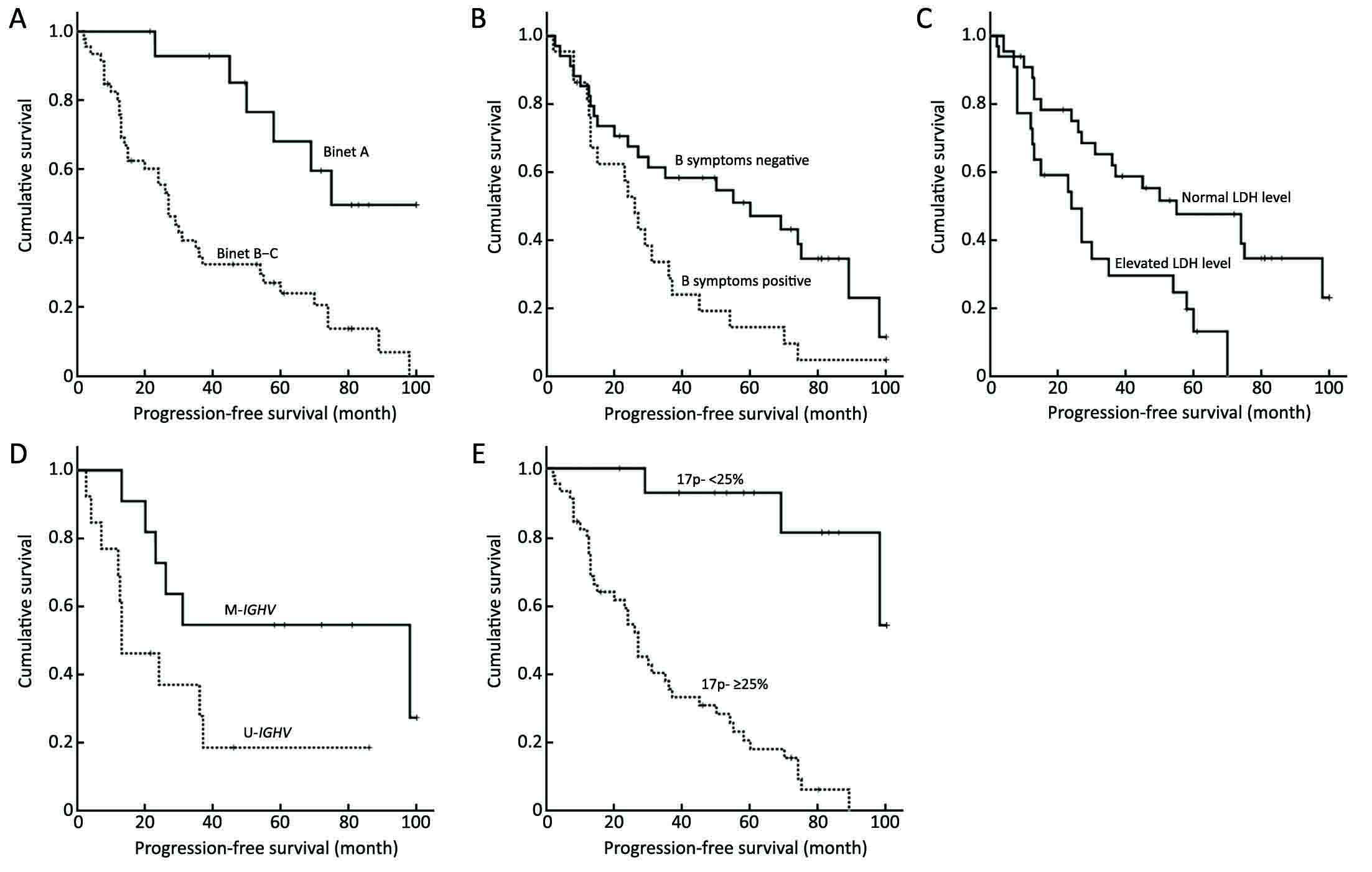
We further assessed the prognostic impact of some clinical and biological variables for disease progression of patients harboring a 17p-. Figure 3 shows the variables significantly associated with a shorter PFS as follows: Binet stage B-C, elevated LDH level, B symptoms, ≥25% of 17p-deleted nuclei and unmutated IGHV status. However, according to the Cox regression analysis, only the LDH concentration was identified as an independent prognostic factor (P=0.012) for PFS (Table 3) within the 17p- CLL group.
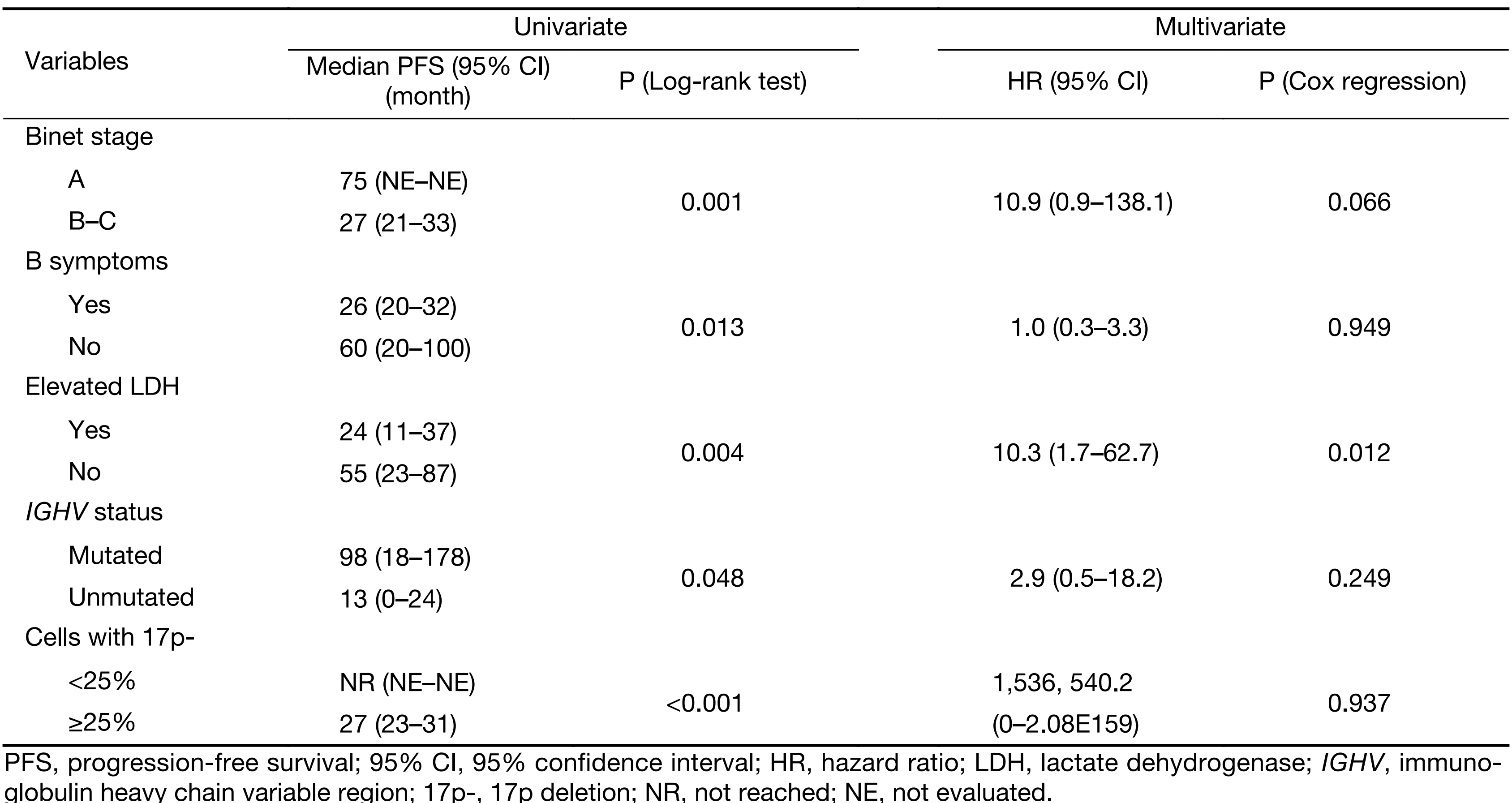
Full table
Discussion
Cytogenetic abnormalities play a critical role in the clinical course of patients with CLL, among which a 17p- and an 11q- have been associated with an unfavorable outcome (3). Our results demonstrated that patients with a 17p- presented a relatively active disease, as suggested by a more advanced clinical stage, higher LDH level, lower median hemoglobin level and higher proportion of patients with B symptoms and unmutated IGHV, which is in concordance with previously published data (4). In addition, a 17p- was significantly associated with worse survival, in terms of TTFT, PFS and OS, which could be due to the deregulation of the cell cycle because of the loss of gene TP53 (16).
In our single-institute cohort, a 17p- was detected by FISH in 14.5% of newly diagnosed patients. The incidence is consistent with another Chinese study (16%) (17) and studies in other countries [22.9% in Czech (18), 18.3% in Spain (19)]. Regarding the optimal cut-off value of the 17p-, there have been some controversies among different studies. Döhner et al. (5) used 3% as the cut-off value, whereas in the CLL4 trial, the cut-off point was set at 20% (20), and the final cut-off value was set at 10% because the outcome of cases with a 10%–20% loss was similar to that of patients with more than 20% deletion in the CLL4 trial (21). According to our study, patients with ≥25% deleted nuclei were considered to be carrying 17p-, which is in line with several other studies (11,19).
The standard treatment regimen for “fit” patients with CLL is based on FC or FCR regimens. Chemotherapy based on fludarabine was reported by several studies to be not very efficacious in patients carrying a 17p- (20,22) because the activity was dependent on the TP53 pathway. According to a systematic review by the CLL Trialists’ Collaborative Group (CLLTCG), FC regimen could improve PFS of patients with CLL compared with regimens without purine analogues, but there was no major difference in the subgroup analysis of the presence/absence of a 17p- concerning the quantity, quality and duration of responses (23). In general, the addition of rituximab to the FC regimen as a frontline therapy in the 17p- population is superior in terms of ORR and PFS (24,25). Alemtuzumab (26) or high-dose corticosteroids in combination with rituximab (R-HDMP) (27) has been suggested as frontline therapies in patients with 17p-, as a higher ORR was shown in the population following these regimens. However, the median PFS remains unsatisfactory (28). Based on the evidence at that time of day, only allogeneic stem cell transplantation holds the prospect for better survival (29). Recently, a wide spectrum of novel drugs acting independently of TP53 have been tested, including ibrutinib [Bruton’s tyrosine kinase (BTK) inhibitor] (30), idelalisib (inhibitor of phosphatidylinositol 3-kinase) (31,32), and ABT-199 (Bcl-2 inhibitor) (33). Given the significant activity of those drugs, the necessity of allogenic stem cell transplantation in patients with the 17p- or TP53 mutation may be reconsidered (34), but most guidelines still include allo-transplantation since it is the only curative treatment (35). In our study, 45.1% of the patients enrolled received alkylating agent-based regimens other than the purine analogue, since they are relative affordable in developing countries compared to rituximab. Four CRs and three PRs were recorded among the eight patients who received FCR, and the ORR was 100% among patients treated with R-HDMP or alemtuzumab. Regarding the clone size of 17p-, the ORR and TTP were inferior in patients with high percentage 17p- compared with low percentage 17p-, which calls for a new therapeutic strategy for treatment of this aggressive disease, especially for those with high percentage 17p-.
Except for the general aggressiveness of the clinical course of patients harboring 17p-, there remains heterogeneity within the subgroup. In the present study, Binet stage A patients with 17p- and mutated IGHV have a relative favorable outcome, which is in consistent with another study (36), indicating the necessity to take different therapeutic strategies depending on the disease status. Further, LDH, one of the most common serum markers, acted as an independent predictor for PFS according to the multivariate analyses, with a median PFS of only two years in patients with both 17p- and a high level of LDH, which was not surprising given that the prognostic value of LDH was reported in many other studies (37). However, its additional significance in 17p- CLL patients is to our knowledge reported here for the first time. Nevertheless, these findings are based on a single-centre retrospective study and the predictive effect was not evaluated. Further studies with relatively larger numbers of patients and longer follow-up periods should be undertaken to confirm our preliminary results.
As a limitation of our study, TP53-mutation detection was not commonly available within this cohort, which was reported to act as a driver of mutation and result in a poor survival for patients with CLL even as a small subclone (38).
Conclusions
Patients positive for a 17p- experience an aggressive clinical course, yet are still heterogeneous. In addition, we agree that the FISH approach should be conducted in all treatment-naive patients. It is necessary to further refine the treatment for patients carrying this aberration, and clinical trials should be taken into consideration when treating this subgroup of patients.
Acknowledgements
This work is supported by grants from the National Nature Science Foundation of China (No. 81200395, 81370632), the National Science and Technology supporting Program (No. 2014BAI09B12), the Fundamental Application and Advanced Technology Research Program of Tianjin (No. 15JCYBJC27900), and the National Public Health Grand Research Foundation (No. 201202017).
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2013, National Cancer Institute. Bethesda, MD. Available online: http://seer.cancer.gov/csr/1975_2013/
- Chihara D, Ito H, Matsuda T, et al. Differences in incidence and trends of haematological malignancies in Japan and the United States. Br J Haematol 2014;164:536–45. [PubMed] DOI:10.1111/bjh.12659
- Chen C, Puwada S. Prognostic factors for chronic lymphocytic leukemia. Curr Hematol Malig Rep 2016;11:37–42. DOI:10.1007/s11899-015-0294-x
- Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000;343:1910–6. DOI:10.1056/NEJM200012283432602
- Döhner H, Fischer K, Bentz M, et al. p53 gene deletion predicts for poor survival and non-response to therapy with purine analogs in chronic B-cell leukemias. Blood 1995;85:1580–9. [PubMed]
- Foà R, Del Giudice I, Guarini A, et al. Clinical implications of the molecular genetics of chronic lymphocytic leukemia. Haematologica 2013;98:675–85. [PubMed] DOI:10.3324/haematol.2012.069369
- Byrd JC, Smith L, Hackbarth ML, et al. Interphase cytogenetic abnormalities in chronic lymphocytic leukemia may predict response to rituximab. Cancer Res 2003;63:36–8. [PubMed]
- Zenz T, Döhner H, Stilgenbauer S. Genetics and risk-stratified approach to therapy in chronic lymphocytic leukemia. Best Pract Res Clin Haematol 2007;20:439–53. [PubMed] DOI:10.1016/j.beha.2007.02.006
- Lozanski G, Heerema NA, Flinn IW, et al. Alemtuzumab is an effective therapy for chronic lymphocytic leukemia with p53 mutations and deletions. Blood 2004;103:3278–81. [PubMed] DOI:10.1182/blood-2003-10-3729
- Thornton PD, Matutes E, Bosanquet AG, et al. High dose methylprednisolone can induce remissions in CLL patients with p53 abnormalities. Ann Hematol 2003;82:759–65. [PubMed] DOI:10.1007/s00277-003-0710-5
- Tam CS, Shanafelt TD, Wierda WG, et al. De novo deletion 17p13.1 chronic lymphocytic leukemia shows significant clinical heterogeneity: the M. D. Anderson and Mayo Clinic experience. Blood 2009;114:957–64. [PubMed] DOI:10.1182/blood-2009-03-210591
- Harris NL, Jaffe ES, Diebold J, et al. The World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. Report of the Clinical Advisory Committee meeting, Airlie House, Virginia, November, 1997. Ann Oncol 1999;10:1419–32. [PubMed]
- Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008;111:5446–56. [PubMed] DOI:10.1182/blood-2007-06-093906
- An G, Xu Y, Shi L, et al. Chromosome 1q21 gains confer inferior outcomes in multiple myeloma treated with bortezomib but copy number variation and percentage of plasma cells involved have no additional prognostic value. Haematologica 2014;99:353–9. [PubMed] DOI:10.3324/haematol.2013.088211
- An G, Li Z, Tai YT, et al. The impact of clone size on the prognostic value of chromosome aberrations by fluorescence in situ hybridization in multiple myeloma . Clin Cancer Res 2015;21:2148–56. [PubMed] DOI:10.1158/1078-0432.CCR-14-2576
- Rossi D, Rasi S, Spina V, et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood 2013;121:1403–12. [PubMed] DOI:10.1182/blood-2012-09-458265
- Xu W, Li JY, Li L, et al. Fluorescent in situ hybridization with a panel of probes detects molecular cytogenetic abnormalities in patients with chronic lymphocytic leukemia . Zhonghua Yi Xue Za Zhi (in Chinese) 2008;88:2537–40. [PubMed]
- Michalová K, Zemanová Z, Cmunt E, et al. The interphase fluorescence in situ hybridization (I-FISH) technique in patients with chronic lymphatic leukemia (CLL) . Cas Lek Cesk (in Czech) 2000;139:334–8. [PubMed]
- Delgado J, Espinet B, Oliveira AC, et al. Chronic lymphocytic leukaemia with 17p deletion: a retrospective analysis of prognostic factors and therapy results. Br J Haematol 2012;157:67–74. [PubMed] DOI:10.1111/j.1365-2141.2011.09000.x
- Catovsky D, Richards S, Matutes E, et al. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 2007;370:230–9. [PubMed] DOI:10.1016/S0140-6736(07)61125-8
- Oscier D, Wade R, Davis Z, et al. Prognostic factors identified three risk groups in the LRF CLL4 trial, independent of treatment allocation. Haematologica 2010;95:1705–12. [PubMed] DOI:10.3324/haematol.2010.025338
- Grever MR, Lucas DM, Dewald GW, et al. Comprehensive assessment of genetic and molecular features predicting outcome in patients with chronic lymphocytic leukemia: results from the US Intergroup Phase III Trial E2997. J Clin Oncol 2007;25:799–804. [PubMed] DOI:10.1200/JCO.2006.08.3089
- CLL Trialists’ Collaborative Group. Systematic review of purine analog treatment for chronic lymphocytic leukemia: lessons for future trials. Haematologica 2012;97:428–36. [PubMed] DOI:10.3324/haematol.2011.053512
- Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophos-phamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010;376:1164–74. [PubMed] DOI:10.1016/S0140-6736(10)61381-5
- Zenz T, Eichhorst B, Busch R, et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol 2010;28:4473–9. [PubMed] DOI:10.1200/JCO.2009.27.8762
- Parikh SA, Keating MJ, O’Brien S, et al. Frontline chemoimmunotherapy with fludarabine, cyclophos-phamide, alemtuzumab, and rituximab for high-risk chronic lymphocytic leukemia. Blood 2011;118:2062–8. [PubMed] DOI:10.1182/blood-2011-01-329177
- Castro JE, James DF, Sandoval-Sus JD, et al. Rituximab in combination with high-dose methylprednisolone for the treatment of chronic lymphocytic leukemia. Leukemia 2009;23:1779–89. [PubMed] DOI:10.1038/leu.2009.133
- Tam CS, Stilgenbauer S. How best to manage patients with chronic lymphocytic leuekmia with 17p deletion and/or TP53 mutation?. Leuk Lymphoma 2015;56:587–93. [PubMed] DOI:10.3109/10428194.2015.1011641
- Dreger P, Döhner H, Ritgen M, et al. Allogeneic stem cell transplantation provides durable disease control in poor-risk chronic lymphocytic leukemia: long-term clinical and MRD results of the German CLL Study Group CLL3X trial. Blood 2010;116:2438–47. [PubMed] DOI:10.1182/blood-2010-03-275420
- Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013;369:32–42. [PubMed] DOI:10.1056/NEJMoa1215637
- Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med 2014;370:997–1007. [PubMed] DOI:10.1056/NEJMoa1315226
- Brown JR, Byrd JC, Coutre SE, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood 2014;123:3390–7. [PubMed] DOI:10.1182/blood-2013-11-535047
- Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med 2016;374:311–22. [PubMed] DOI:10.1056/NEJMoa1513257
- Jain N, O’Brien S. Chronic lymphocytic leukemia with deletion 17p: emerging treatment options. Oncology (Williston Park) 2012;26:1067,1070. [PubMed]
- Ghielmini M, Vitolo U, Kimby E, et al. ESMO Guidelines consensus conference on malignant lymphoma 2011 part 1: diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL) and chronic lymphocytic leukemia (CLL). Ann Oncol 2013;24:561–76. [PubMed] DOI:10.1093/annonc/mds517
- Best OG, Gardiner AC, Davis ZA, et al. A subset of Binet stage A CLL patients with TP53 abnormalities and mutated IGHV genes have stable disease. Leukemia 2009;23:212–4. [PubMed] DOI:10.1038/leu.2008.260
- Rosenquist R, Cortese D, Bhoi S, et al. Prognostic markers and their clinical applicability in chronic lymphocytic leukemia: where do we stand?. Leu Lymphoma 2013;54:2351–64. [PubMed] DOI:10.3109/10428194.2013.783913
- Rossi D, Khiabanian H, Spina V, et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 2014;123:2139–47. [PubMed] DOI:10.1182/blood-2013-11-539726