
Future of anti-PD-1/PD-L1 applications: Combinations with other therapeutic regimens
Introduction
The programmed cell death 1 (PD-1) pathway has become an attractive therapeutic target in numerous cancers (1). PD-1 was first characterized as a marker of apoptosis, and is now known as an important immune checkpoint that regulates T cell function. PD-1 is widely expressed on tumor-infiltrating lymphocytes (TILs), particularly on exhausted T cells (2). Its ligand, programmed death 1 ligand (PD-L1), is mainly expressed on cancer and immune cells, such as dendritic cells (DCs), macrophages, myeloid-derived suppressor cells (MDSCs), and B cells (3). Mechanistic studies have shown that PD-1/PD-L1 signaling can inhibit the activation of T cells and thus suppress their proliferation, migration, and effector function by delivering a co-inhibitory signal (4-6). It has been reported that CD8+ T cells often become exhausted when entering a suppressive tumor microenvironment (TME) (7), and exhausted CD8+ T cells are characterized by overexpression of multiple inhibitory receptors, such as PD-1, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), T-cell immunoglobulin mucin-3 (TIM-3), and lymphocyte activation gene 3 (LAG-3), as well as impaired effector cytokine production and cytolytic activity, leading to the failure of cancer elimination (8). Antibody-mediated blockade of these inhibitory receptors and their ligands have yielded breakthrough immunotherapies for numerous cancers (9).
Inhibitors targeting PD-1 and PD-L1 have shown impressive anti-tumor effects and clinical benefits in a subset of patients (10). Food and Drug Administration (FDA) has approved various anti-PD-1/PD-L1 monoclonal antibodies (mAbs) for the treatment of metastatic melanoma, renal cell carcinoma (RCC), non-small cell lung cancer (NSCLC), bladder cancer, recurrent or metastatic head and neck cancer, refractory classical Hodgkin lymphoma, microsatellite instability (MSI)-high colorectal cancer (CRC), and metastatic Merkel cell carcinoma. Despite these unprecedented successes, most patients experience intrinsic resistance, and even responding patients can develop acquired resistance to anti-PD-1 therapy (11). Numerous studies are currently underway to systematically elucidate the mechanisms underlying this resistance. Here, we summarize the major resistance mechanisms: 1) T cell exhaustion at the tumor site; 2) exclusion of pre-existing T cells or TILs; 3) the immunosuppressive TME formed by suppressive cells and inhibitory molecules and 4) tumor-intrinsic factors, such as oncogenic signaling pathways, PD-L1 expression levels, tumor immunogenicity, expression of major histocompatibility complex class I (MHC-I) molecules and immunosuppressive metabolites. Therefore, exploring effective combinatorial approaches to recover sensitivity in PD-1 blockade-resistant patients is attracting research directions. Herein, we briefly review the development of PD-1 pathway, discuss the current limitations of anti-PD-1/PD-L1 therapies, and then focus on potential combinatorial strategies, aiming to guide future combination trials that might yield better outcomes for cancer patients.
Discovery of PD-1 and its ligands
In 1992, Ishida et al. isolated the cDNA of PD-1 and identified PD-1 as a marker of classical programmed cell death (apoptosis) (12). Subsequent mechanistic studies identified PD-1 as a negative regulator of T cell immune responses, which also played a role in preventing autoimmune diseases (13). In 2000, B7 homolog 1 (B7-H1, also called PD-L1 or CD274), a member of the B7 family of costimulatory molecules, was confirmed as a specific ligand of PD-1 (14). Similar to PD-1-deficient mice, PD-L1-deficient mice exhibited immune activation and autoimmune diseases (14). PD-L1 was shown to deliver inhibitory signals by binding to CD80 on activated T cells (15). In addition to PD-L1, programmed death ligand 2 (PD-L2) was also demonstrated to be a second ligand of PD-1 that is expressed by DCs, macrophages, and some cancer cells. Interaction of PD-L2 with PD-1 also mediated potent inhibitory signals to hinder the proliferation and function of T effector cells (16). PD-L2 was also reported to be involved in inducing respiratory tolerance through interaction with repulsive guidance molecule family member b (RGMb), which is highly expressed on lung macrophages (17). Collectively, interaction of PD-L1 with PD-1 suppresses the initial activation and effector function of T cells, and blocking PD-1/PD-L1 pathway would enhance T cell immune responses.
Interaction of PD-1 with PD-L1 in the TME
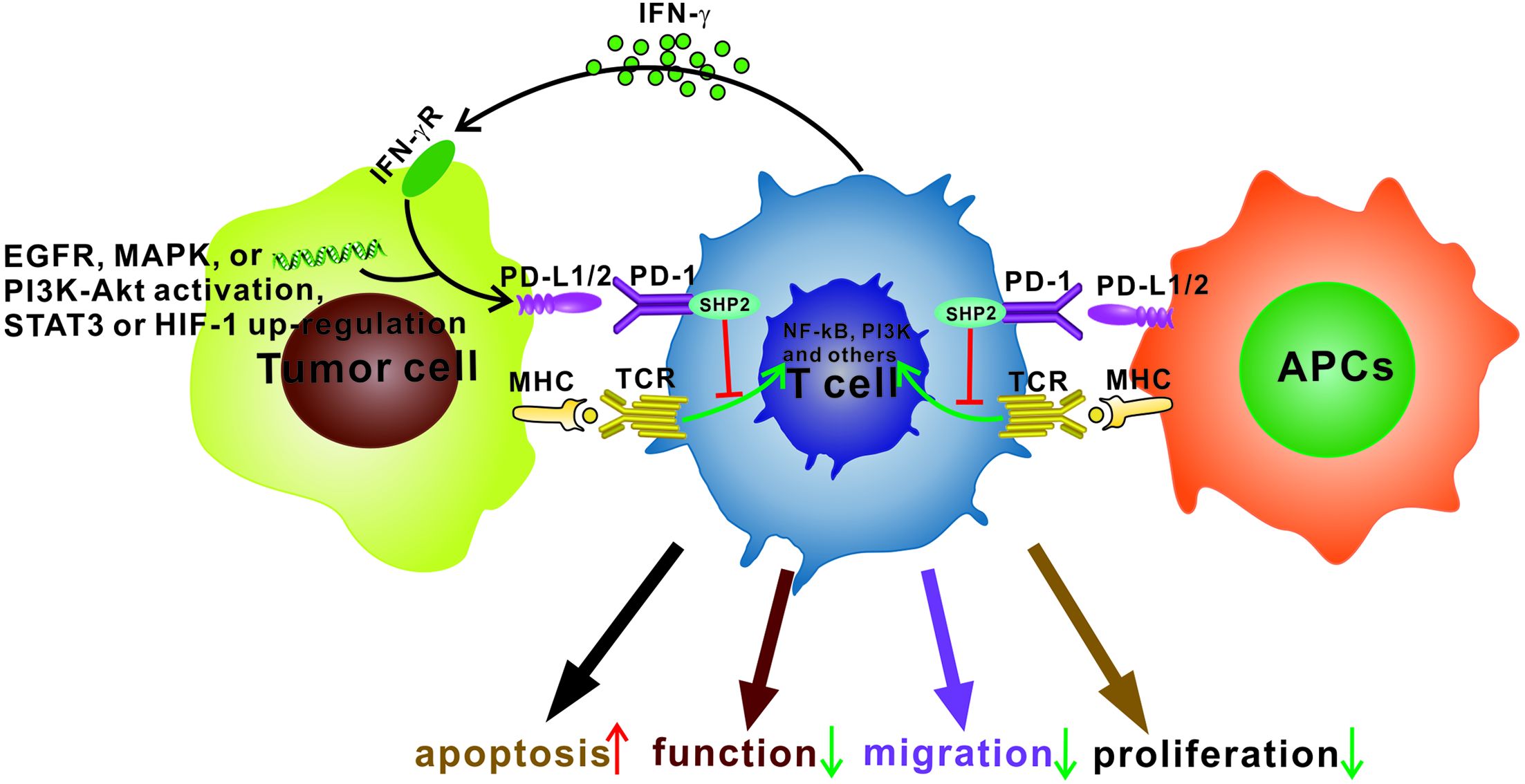
In 2002, Dong et al. first reported the role of PD-1 pathway in promoting T cell apoptosis in the TME (18). Subsequent studies demonstrated that interaction of PD-1 with PD-L1 attenuated T cell migration, proliferation, and cytotoxic capacity (4-6) (Figure 1). Interestingly, a recent study revealed a novel role of PD-1 in cancer progression; PD-1-positive tumor-associated macrophages (TAMs) could attenuate tumor immunity by inhibiting phagocytosis (19). The major mechanism by which PD-1/PD-L1 limited the host immune response was through upregulation of their expression in the TME, and elevated PD-1 expression was detected on TILs, particularly tumor antigen-specific T cells, which was presumably induced by chronic antigenic stimulation (2). PD-L1 can be highly expressed by tumor cells and tumor-associated antigen-presenting cells (APCs) (3), and the levels of PD-L1 expressed by tumor cells can be regulated by various mechanisms (Figure 1). One of these mechanisms may involve inflammatory cytokines, especially interferon-γ (IFN-γ) (20). Another mechanism is aberrant expression of tumor-intrinsic signaling pathways during carcinogenesis, including activation of epidermal growth factor receptor (EGFR), mitogen-activated protein kinase (MAPK), and/or PI3K-Akt signaling pathways, and upregulation of signal transducer and activator of transcription 3 (STAT3) or hypoxia induced factor 1 (HIF-1) (21,22). PD-L1-positive cells not only inhibited T cell survival and function but also improved the suppressive activity of DCs (23) and T regulatory cells (Tregs) (24). Additionally, PD-L1 delivered an anti-apoptotic signal to tumor cells through its interaction with PD-1 and protected tumor cells from attack by cytotoxic T lymphocytes (CTLs) (25). PD-1/PD-L1 blockade has been shown to recover the survival and function of T cells and increase their tumor migration, significantly reinvigorating their anti-tumor immunity (26,27). In summary, PD-1/PD-L1 pathway could be used as a therapeutic target based on its immunosuppressive role in the TME, and thus modulating PD-1/PD-L1 pathway could remodel the immunosuppressive microenvironment.
Anti-PD-1/PD-L1 agents
Anti-PD-1/PD-L1 mAbs augment T-cell anti-tumor response by blocking PD-1/PD-L1 interaction. FDA has approved the anti-PD-1 mAb nivolumab for advanced melanoma, squamous cell carcinoma, metastatic NSCLC, advanced kidney cancer, Hodgkin lymphoma, and urothelial carcinoma. Pembrolizumab, another anti-PD-1 mAb, was approved by FDA for the treatment of unresectable or metastatic melanoma, advanced NSCLC, and MSI-high CRC. In 2016, the anti-PD-L1 mAb MPDL3280A was approved by FDA for the treatment of advanced or metastatic urothelial bladder cancer (UBC). Clinical trials of another PD-L1 mAb, BMS-936559, have also been conducted for various advanced cancers (28). The immune-related adverse events (irAEs) of these anti-PD-1/PD-L1 mAbs were mostly consistent, including fatigue, asthenia, pyrexia, myalgia, arthralgia, rash, pruritus, nausea, vomiting, colitis, liver and kidney toxicity, pneumonia, dyspnea, anemia, injection-related reactions, and endocrine disorders.
Limitations of anti-PD-1/PD-L1 therapy
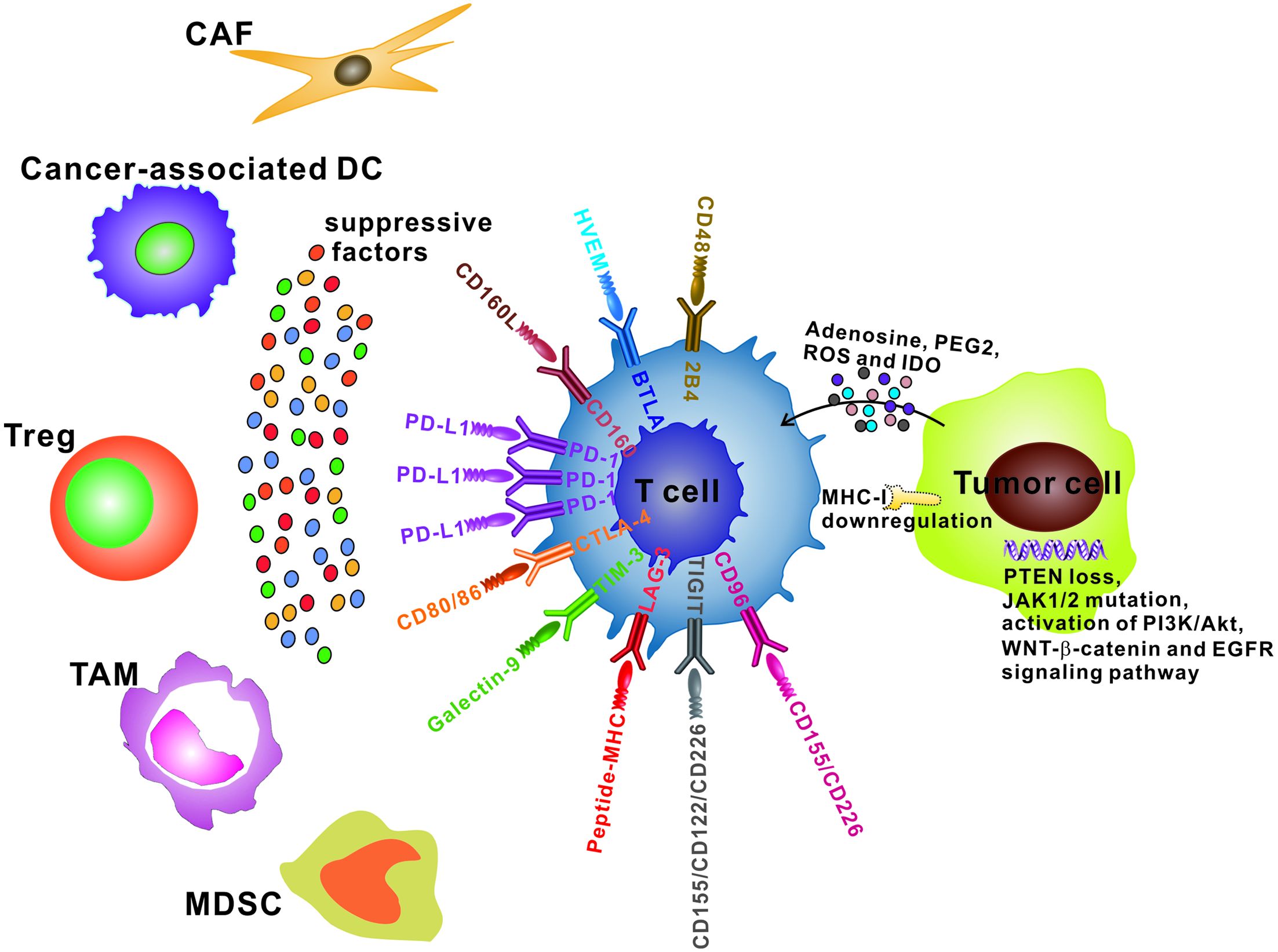
Despite these encouraging clinical results, anti-PD-1/PD-L1 agents are not always effective. The majority of patients did not benefit from anti-PD-1/PD-L1 therapy (primary resistance), some responders relapsed after a period of response (acquired resistance) (11), and some patients had to stop treatment because of the development of irAEs. Here, we summarize the known mechanisms promoting resistance to anti-PD-1/PD-L1 therapy (Figure 2). First, emerging evidence shows that decreased therapeutic efficacy can be caused by T cell exhaustion. Upregulation of alternative immune checkpoints or a lack of effector and memory epigenetic profile resulted in T cell re-exhaustion after PD-1 blockade, limiting the durability of reinvigoration (29,30). Ngiow et al. analyzed the expression of PD-1 by intratumor CD8+ T cells in anti-PD-1 sensitive and resistant tumors and identified a threshold for PD-1 expression, below which, release of adaptive immune resistance could be achieved with anti-PD-1 mAb, whereas T cells with expressing PD-1 levels above the threshold were in severe exhausted status and failed to be rescued by anti-PD-1 therapy (31). As a second mechanism, exclusion of pre-existing T cells or inadequate T cell trafficking to the tumor site was identified as a key factor contributing to resistance to PD-1/PD-L1 blockade. It was shown that sensitivity to anti-PD-1/PD-L1 therapy required efficient trafficking of pre-existing CD8+ T cells or effector T cells (Teffs) to the TME (32). Third, many suppressive factors were shown to accumulate in the TME, including Tregs, MDSCs, TAMs, immature DCs, and cytokines, such as interleukin 6 (IL-6) (33), interleukin 10 (IL-10) (34), and vascular endothelial growth factor A (VEGF-A) (35), which affected the clinical efficacy of PD-1/PD-L1 blockade. In addition, many tumor-intrinsic factors were also extensively associated with the failure of anti-PD-1/PD-L1 therapy, including upregulation of oncogenic signaling pathways (36), expression levels of PD-L1 (37), tumor immunogenicity (3), expression of MHC-I (38), and tumor suppressive metabolites, such as indoleamine 2,3-dioxygenase (IDO) (39) and adenosine (40). Recently, a novel explanation for therapeutic resistance to anti-PD-1/PD-L1 therapy was proposed, in which, PD-1+ TAMs capture anti-PD-1 mAbs on T cell surface through specific Fc/Fcγ receptor interactions, and blocking Fcγ receptor before anti-PD-1 mAb administration prolonged the binding of anti-PD-1 mAb to CD8+ TILs and improved the tumoricidal effect in mice (41). Taken together, combinatorial therapies targeting the PD-1/PD-L1 pathway and the mechanisms of resistance provide a rationale for sensitizing the resistant patients.
Potential combinatorial strategies with PD-1/PD-L1 blockade
Based on the limited therapeutic effect of single anti-PD-1/PD-L1 therapy, it is urgent to explore effective combinatorial approaches to overcome anti-PD-1/PD-L1 therapy resistance and provide insights into clinical applications. Potential therapeutic combinations with PD-1/PD-L1 blockade are shown in Figure 3. In this section, the potential therapeutic regimes synergizing with PD-1/PD-L1 blockade are discussed.

Chemotherapy
Some chemotherapeutic agents treat cancers in part by enhancing anti-tumor immunity in a subset of patients by eliminating Tregs and enhancing CD8+ T cell responses (42). However, in some cancer patients, chemotherapeutic agent-induced anti-tumor immunity prompted PD-L1 expression to foster an immunosuppressive TME (43). Activation of PD-1/PD-L1 pathway was reported to confer tumor cell chemoresistance associated with increased metastasis (44). Preclinical and clinical studies have confirmed that chemotherapy combined with anti-PD-1 mAb synergistically facilitated anti-tumor immune response (45) and improved survival in a subset of patients (46). In 2017, the American Society of Clinical Oncology (ASCO) reported that the disease control rate of anti-PD-1 mAb combined with chemotherapy for colon and gastric cancer was up to 100% (47) and 92% (48), respectively. The details of how chemotherapy, including the agents, dose and timing, should be used in combination with anti-PD-1/PD-L1 mAbs warrant further exploration in patients with different types of cancer.
Radiation therapy (RT)
High-dose ionizing radiation has been reported to eliminate tumor cells partially depending on anti-tumor immunity, particularly T cell immune response. Radiation functions by upregulating MHC-I expression and tumor-associated antigen expression, thus enhancing T cell migration, activating DCs, and diminishing the accumulation of Tregs (49). However, RT rarely resulted in tumor regression at sites distant to the irradiated field, an immune-mediated response termed the “abscopal effect” (50). Preclinical studies showed that RT-induced PD-L1 expression might compromise the anti-tumor effect and lead to local relapse after RT (51). However, the combination of anti-PD-1/PD-L1 mAbs with RT was reported to augment the abscopal effect and promote T cell anti-tumor immunity in several tumor-bearing mouse models (51,52). Encouraging clinical outcomes have been observed in patients with metastatic melanoma treated with RT and anti-PD-1 therapy (53). A new additional target of this combinatorial therapy is the GAS6/AXL pathway, and targeting this pathway resulted in greater anti-tumor immune responses after RT in some cancers (54). Based on these findings, RT could be used as an adjuvant approach for anti-PD-1/PD-L1 therapy. However, to translate such combinatorial therapies into the clinic, further preclinical and clinical studies are required to detect the ionizing radiation-induced immunogenic factors and optimize the agents, dose, schedule, location and opportunity.
Targeting T cell exhaustion and exclusion
Adoptive T cell transfusion (ACT)
Accumulating evidence has shown that exclusion of effector T cells leads to the failure of cancer regression in most patients, and ACT was considered as a personalized therapy that involved administration to the host of highly tumor-reactive T cells (55). Various ACT therapies, such as chimeric antigen receptor (CAR)-T cell and T cell receptor (TCR)-T cell transfusion, have shown great clinical benefit in hematological and solid tumors (56). However, many patients do not response because of the limited factors, such as inadequate T cell trafficking, high expression levels of inhibitory receptors, tumor metabolism and the immunosuppressive TME (57). PD-1 was found to be upregulated on the transferred TILs, leading to their dysfunction. However, co-administration of adoptive T cells with an anti-PD-1 mAb reinvigorated exhausted TILs and strengthened anti-tumor immunity in lung tumor-bearing mice (57). A recent study showed that PD-1/PD-L1-induced resistance compromises the efficacy of CAR-T cell therapies in solid tumors, and administration of anti-PD-1 mAb, cell-intrinsic PD-1 shRNA blockade, or a PD-1 dominant-negative receptor restored the effector function of CD28 CAR-T cells (58). These findings suggest PD-1/PD-L1 blockade as a potential strategy to enhance the potency of ACT therapies. Several clinical studies of combinations of TCR-T cell or CAR-T cell transfusion with anti-PD-1/PD-L1 mAbs are ongoing (NCT02858310, NCT02775292, NCT03030001, NCT02873390, NCT02862028, and NCT02926833).
Targeting co-inhibitory molecules on T cells
Apart from PD-1, other checkpoint molecules, such as CTLA-4 (59), TIM-3 (60), LAG-3 (61), and T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT) (62), were also upregulated by exhausted T cells in some cancer patients. Co-expression of these inhibitory receptors probably aggravated the severity of T cell exhaustion, and combined blockade of these inhibitory molecules is an inspiring strategy for cancer therapy. It has been reported that CTLA-4 blockade not only enhanced the effector function of T cells but also eliminated Tregs and MDSCs in the TME (63,64). Clinical trials of combinatorial treatments with anti-CTLA-4 and anti-PD-1 agents have demonstrated promising clinical results in patients with advanced metastatic tumors, particularly melanoma (65). FDA approved this combinatorial therapy for the treatment of BRAF V600 wild-type unresectable or metastatic melanoma. However, superimposed toxicity, abnormal response patterns, and other serious adverse events were observed. Thus, sequential treatment with anti-PD-1/PD-L1 mAbs followed by anti-CTLA-4 mAbs needs to be carefully considered.
Combinatorial targeting of PD-1 and the TIM-3 (60), LAG-3 (61) and TIGIT (62) was shown to synergistically recover exhausted T cells and enhance anti-tumor effects in some types of cancer. A new triple therapy of anti-TIM-3, anti-PD-1, and radiation for human glioblastoma showed 100% overall survival (OS) (66). The safety and efficacy of dual PD-1 and TIM-3 blockade along with cancer vaccines in patients with advanced melanoma have been tested in a phase I clinical trial (67). In addition, several novel co-inhibitory receptors involved in CD8+ T cell exhaustion have been identified, such as CD96 (68), 2B4 (CD244) (69), CD160 (70), and B- and T-lymphocyte attenuator (BTLA) (71), and blocking them could partially restore T cell function and/or expansion and might enhance anti-tumor immunity (69-71). Collectively, combinatorial targeting PD-1/PD-L1 and alternative checkpoints on T cells have great potential for T-cell based immunotherapy, and several clinical trials are ongoing (NCT02817633, NCT02608268, NCT02658981, NCT01968109, NCT02061761, NCT03005782, NCT02676869, NCT02966548, and NCT02460224).
Activating co-stimulatory molecules on T cells
4-1BB (CD137, TNFRSF9), OX40, CD27 and CD40 are members of tumor necrosis factor receptor (TNFR) superfamily, which provide strong costimulatory signals for augmenting and diversifying T-cell responses (72). Tumor-reactive CD8+ T cells frequently co-express PD-1 and costimulatory molecules of the TNFR family (73), offering a repertoire of potential targets for T cell-based therapy.
4-1BB has been identified as a potent mitigator of exhausted T cells, and administration of an anti-4-1BB agonist significantly enhanced CAR-T-cell efficacy in solid tumor settings (74). Preclinical studies have shown that 4-1BB activation and PD-1 blockade synergistically promoted T-cell anti-tumor responses in a mouse model of poorly immunogenic tumors (75). In addition, a recent study revealed that co-treatment with anti-4-1BB agonist, anti-PD-L1 mAb, and a tumor vaccine produced more powerful anti-tumor capacity by reprogramming suppressive and stimulatory signals in ovarian cancer (76). Clinical trials of this combinatorial therapy for the treatment of advanced solid tumors are ongoing (NCT02179918, NCT02652455, NCT02845323, and NCT02554812). Unfortunately, severe liver toxicity has become the biggest obstacle for the clinical application of anti-4-1BB agonists.
Both OX40 and CD27 costimulatory signals could reinvigorate exhausted CD8+ T cells. Anti-PD-1/OX40 mAb treatment was shown to increase CD4+ T and CD8+ T cells and decrease Tregs and MDSCs (77). Either OX40 or CD27 agonistic mAb combined with anti-PD-L1 mAb synergistically enforced CD8+ T cell proliferation and effector cytokine generation in mice (78). The ongoing clinical trials are as follows: combination of third generation CAR-T cells, iC9-GD2-CD28-OX40 (iC9-GD2) T cells, with anti-PD-1 mAb in relapsed or refractory neuroblastoma (NCT01822652); and combination of agonistic CD27 mAb and anti-PD-1 mAb in advanced refractory solid tumors (NCT02335918).
It has been reported that CD40 activation rescued antiviral CD8+ T cells from PD-1-mediated exhaustion (79). Combination of agonistic CD40 mAb with chemotherapy completely reversed the resistance to anti-PD-1 mAb in pancreatic tumors (80), and a clinical trial of nivolumab in combination with GM. CD40L vaccine in adenocarcinoma of the lung is ongoing (NCT02466568). However, the appropriate dose, schedule, route of administration, and formulation for these combinatorial therapies need to be determined for treating different cancer types.
Modulating T cell metabolism
It has been reported that a hostile TME can restrain T-cell immune response by altering cellular metabolism, such as impaired glycolysis (81,82), fatty acid metabolic dysregulation (83,84), aberrant cholesterol esterification (85), disruption of mitochondrial function, and the generation of high levels of reactive oxygen species (ROS) (86). Hypoxia-mediated glycolysis was shown to interfere with the differentiation and function of T cells by modulating the expression of co-inhibitory receptors, including PD-1, CTLA-4, and LAG-3 (87). However, anti-PD-1/PD-L1 mAbs could partially restore T cell glycolysis and function (81). One mechanism by which PD-1 inhibits T-effector cell differentiation is through the enhancement of fatty acid-oxidation (FAO) of endogenous lipids (83). Free linoleic acid has been reported to cause T cell loss by ROS-mediated depletion and promote liver carcinogenesis (83). Additionally, limiting cholesterol esterification in T cells through inhibition of acetyl-coenzyme A acetyltransferase 1 (ACAT1), which is a key cholesterol esterification enzyme, enhanced the proliferation and effector function of CD8+ T cells, and combination treatment with ACAT1 inhibitors and anti-PD-1 mAb demonstrated remarkable tumor regression in mice with melanoma (85). In-depth investigations of the association between PD-1 expression and metabolism in dysfunctional T cells are needed to guide anti-PD-1 therapy.
Promoting T-effector cell infiltration
Chemokines play a role as chemoattractants of different leukocyte subsets and act as a double-edged sword in tumor development. For example, Th1-type chemokines C-X-C Motif Chemokine Ligand 9 (CXCL9) and C-X-C Motif Chemokine Ligand 10 (CXCL10) can increase effector T cell trafficking. It has been verified that upregulation of CXCL9 and CXCL10 improved the therapeutic efficacy of anti-PD-1 mAb (88). However, it also has been recently reported that adenosine signaling in the TME suppressed CXCL9 and CXCL10 production and T-cell infiltration in metastatic lung cancer, and this suppression could be partially reversed by adenosine receptor antagonist (89). Therefore, targeting Th1-type chemokines might offer an effective approach to combinatorial therapy with anti-PD-1/PD-L1 mAb, particularly in tumors with poor T-cell infiltration. In addition, the immunosuppressive stromal cell derived factor-1 (SDF1α)/CXCR4 axis was found to be upregulated in sorafenib-treated advanced hepatocellular carcinoma, which enhanced the expression of PD-L1 and the recruitment of Tregs and M2-type macrophages (90,91). Blockade of SDF1α/CXCR4 axis had no significant effect on CTL infiltration and function but altered the suppressive state of the TME. Dual blockade of PD-1 and CXCR4 boosted CD8+ TIL responses and prevented the polarization toward an immunosuppressive TME in mice (91), providing a rationale for combinatorial blockade of PD-1 and SDF1/CXCR4 axis in clinical studies.
Targeting immunosuppressive factors in the TME
The TME consists of cancer cells, inflammatory cells, stromal cells, and cytokines. These components form a complicated immunosuppressive network that limits T-cell activation and induces T-cell dysfunction (Figure 2). The immunosuppressive nature of the TME has emerged as a critical regulator of anti-tumor immune responses, and it is increasingly being recognized as a major barrier to the effectiveness of cancer immunotherapy. Some tumors resistant to anti-PD-1/PD-L1 therapy have been shown to simultaneously utilize multiple immunosuppressive pathways in the TME, and these pathways are described below.
Suppressive cytokines
IL-10 is produced by multiple cells in the TME, and its receptor, IL-10R, was found to be upregulated on activated CD8+ T cells in some cancer patients (92). IL-10/IL-10R interaction was shown to directly limit CD8+ T cell proliferation and survival (93). PD-1 blockade augmented IL-10R expression on tumor antigen-specific CD8+ T cells, and dual PD-1 and IL-10 blockade significantly enhanced the response of antigen-specific CD8+ T cells in vitro (34), offering a rationale for blocking both IL-10 and PD-1 to strengthen T cell anti-tumor activity. In some cancer patients, IL-6 levels were also elevated and closely related to an aggressive cancer phenotype (94). Cancer-associated fibroblast (CAF)-derived IL-6 has been reported to recruit MDSCs and impair TIL function by upregulating inhibitory immune checkpoints, and IL-6 blockade reversed anti-PD-L1 resistance in a hepatocellular carcinoma tumor model (95). Another suppressive cytokine in the TME, transforming growth factor-β (TGF-β), was shown to directly inhibit CD8+ T cell response, promote Treg development, limit DC maturation, and upregulate PD-1 expression on T cells (95-97). It has been recently shown that TGF-inhibition reversed PD-1-induced immune tolerance and reinforced T cell responses (98). Thus, combinatorial administration of TGF-β/TGF-β receptor inhibitors and anti-PD-1/PD-L1 mAbs could be considered as a potential therapy to enhance T cell anti-tumor immunity in cancer immunotherapy.
VEGF-A/VEGF receptor 1 (VEGFR1)
VEGF-A produced by tumor cells and associated immune cells was identified to limit DCs maturation, enhance Tregs and MDSCs accumulation, and upregulate PD-1 expression by stimulating VEGFR1 (35). Anti-VEGF antibody was approved by the FDA as the first antiangiogenic agent several years ago. A clinical study demonstrated that increased levels of PD-L1 were associated with shorter survival in patients with metastatic RCC receiving VEGF-targeted agents (99). Anti-angiogenic agents targeting VEGF-A-VEGFR1 pathway have been reported to dramatically enhance the efficacy of anti-PD-1/PD-L1 therapy in a mouse model of colorectal cancer (100), suggesting that combined targeting VEGF-A-VEGFR and PD-1/PD-L1 pathway might be a potential strategy for VEGF-expressing tumors. However, potent VEGF inhibition might increase tumor hypoxia, which would decrease the therapeutic effect. Therefore, further studies are needed to determine the proper dose titration when combining anti-VEGF therapy with anti-PD-1/PD-L1 therapy.
PI3-kinase γ (PI3Kγ) signaling in myeloid cells and macrophages
Recently, macrophage PI3Kγ was identified as a molecular switch that controlled immune suppression during inflammation and cancer (101), and high PI3Kγ expression increased the suppressive activity of macrophages and infiltrating myeloid cells, which mediated resistance to anti-PD-1/PD-L1 therapy in mouse models of cancer (101,102). Conversely, it was shown that selective inactivation of PI3Kγ could reshape the TME and promote cytotoxic-T-cell-mediated tumor regression, thus improving the therapeutic effect of anti-PD-1/PD-L1 therapy (101,102). These results provide evidence for a combinatorial approach using PI3Kγ inhibitors to overcome resistance to anti-PD-1/PD-L1 therapy in patients with accumulated suppressive myeloid cells or macrophages.
Targeting tumor-intrinsic factors
In addition to T cell autonomous factors and the suppressive TME, tumor-intrinsic factors, such as abnormal tumor metabolism, elevated oncogenic pathways, and loss of tumor antigens, might be potential therapeutic targets in combination with PD-1/PD-L1 blockade.
Targeting immunosuppressive tumor metabolites
Some tumors that are resistant to anti-PD-1 therapy have been shown to utilize multiple tumor metabolites involved in immunosuppression. For example, to survive in a hypoxic environment, cancer cells have been shown to alter purine metabolism, and this alteration also increased CD73 expression by lymphocytes, endothelial, and epithelial cells (103). High levels of CD73 induced the production of extracellular adenosines, which further increased PD-1 expression and Treg accumulation by binding to A2A adenosine receptor, thus promoting resistance to anti-PD-1 therapy (104). Dual CD73 and PD-1 blockade demonstrated synergistic anti-tumor activity in murine tumor models (40). Tumor cell-derived prostaglandin E2 (PGE2) and its synthesizing enzyme, cyclooxygenase-2 (COX-2), serve as both pro-inflammatory mediators and potent immune suppressors of anti-tumor immunity (105,106). Single PD-1 blockade enhanced the production of PGE2 and pro-tumor inflammatory cytokines, attenuating its therapeutic effect, while combinatorial inhibition of PD-1 and PGE2 significantly augmented the function and survival of CTLs during chronic viral infection (107). In addition, dual COX-2 inhibition and PD-1 blockade synergistically improved T cell anti-tumor immunity in mouse model of melanoma (106). Upregulation of IDO has also been detected in many tumors and has been shown to render experimental mouse models of melanoma resistant to anti-PD-1 therapy (39). Targeting IDO-mediated immunosuppression has demonstrated promising synergistic effects when combined with anti-PD-1/PD-L1 mAbs (108). In addition, high levels of ROS, a kind of short-lived, chemically highly reactive and oxygen-containing small molecules, have been reported to suppress the T cell immune responses. ROS could be produced by cancer cells and multiple immunosuppressive cells in the TME (86). PD-1 was shown to alter the lipid metabolism of antigen-specific T cells, which might further promote ROS production and impair their anti-tumor capacity (83,84). These results suggest that targeting the immunosuppressive metabolites of tumor cells might be an effective adjunct therapy to improve the therapeutic effect of PD-1/PD-L1 blockade.
Targeting oncogenic signaling pathways
Many tumor-intrinsic oncogenic signalings are being increasingly recognized as contributors to immune suppression, which affects the efficacy of anti-PD-1/PD-L1 therapy. It has been reported that PTEN loss and PI3K-Akt pathway activation not only directly boost tumor cell growth but also suppress anti-tumor leukocyte recruitment and function in multiple cancers. It was also shown that PI3K inhibition could improve anti-PD-1 mAb activity in murine models (109,110). Stabilization of WNT-β-catenin signaling was identified as an important mechanism of T cell exclusion from cancers by inhibiting CD103+ DC recruitment, which eventually mediated resistance to anti-PD-L1 therapy (111). Activation of EGFR pathway was reported to correlate with the upregulation of PD-1, PD-L1, CTLA-4, and pro-tumor inflammatory cytokines, which inhibited the therapeutic effect of PD-1 blockade (112). In patients with melanoma, JAK1/2 loss-of-function mutations in tumor cells can mediate primary resistance to PD-1 blockade via a genetic lack of reactive PD-L1 expression (36). In addition, tumor cell downregulation of MHC-I molecules represents another common mechanism of tumor immune escape and resistance to anti-PD-1/PD-L1 therapy (38). Taken together, these findings demonstrate that tumor-specific oncogenic signals play important roles in the development of resistance to anti-PD-1/PD-L1 therapy, and targeting them might be an effective approach to enhance the therapeutic effect in some cancer patients.
Targeting mutant tumor neoantigens
Effective anti-PD-1/PD-L1 therapy requires tumor cell recognition by tumor antigen-specific T cells in the TME. This suggests that poorly immunogenic tumors should be largely resistant to anti-PD-1 therapy. However, it has been revealed that tumor neoantigen burden is closely correlated with the immunogenicity and determines the sensitivity to anti-PD-1 therapy (113). Neoantigen-enriched tumors also showed a higher accumulation of CD8+ T cells (114), and PD-1/PD-L1 blockade has been reported to potentiate neoantigen-specific T cell responses (113). MSI-H cancer, resulting from defective mismatch repair proteins (dMMR), is a type of “hot” tumor characterized by high mutation burdens, active immune TME, and high expression levels of immune checkpoints (115). In 2017, FDA approved pembrolizumab for the treatment of MSI-H/dMMR-subtype solid tumors. A recent study demonstrated that immunodominant T cell reactivities were directed against mutant neoantigens or a cancer germline antigen, rather than the canonical antigens in human papillomavirus (HPV)-associated cervical cancer (116). Therefore, the mutant neoantigens in a cancer might predict its sensitivity to anti-PD-1/PD-L1 therapy, and these neoantigens could be targeted in novel combination approach with anti-PD-1/PD-L1 therapy.
Clinical trials of anti-PD-1/PD-L1 mAbs
Anti-PD-1/PD-L1 mAbs have shown encouraging clinical results in both solid tumors and hematological malignancies (Table 1), and clinical trials of some combinatorial therapies are shown in Table 2. These results remind us the great potential of anti-PD-1/PD-L1 mAbs in treating patients.

Full table

Full table
Conclusions
PD-1/PD-L1 blockade represents an attractive therapeutic strategy in cancer immunotherapy. However, primary and adaptive resistance to anti-PD-1/PD-L1 therapy has been reported to occur in 30%–60% of cancer patients. Therefore, a comprehensive understanding of the resistance mechanisms and how to overcome them are critical for improving clinical responses. Combined therapies of PD-1/PD-L1 blockade with adjunctive strategies, such as chemotherapy, RT, ACT, and other immune checkpoint inhibitors, have shown promising succcess by improving the probability, duration, and potency of clinical activity. However, the combinations discussed here require additional research to identify the safety, efficacy and optimal dosage and timing. Additionally, as the microenvironment where immune cells interact with cancer cells is complicated and dynamic, improved understanding of the mechanisms limiting anti-PD-1/PD-L1 therapy is needed to broaden its clinical applicability in the future.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No. 81171986), and the Ministry of Public Health (No. 201501004).
Footnote
Conflicts of Interest: The authors have no conflicts of interests to declare.
References
- Sznol M, Chen L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin Cancer Res 2013;19:1021–34. [PubMed] DOI:10.1158/1078-0432.CCR-12-2063
- Tan J, Chen S, Lu Y, et al. Higher PD-1 expression concurrent with exhausted CD8+ T cells in patients with de novo acute myeloid leukemia . Chin J Cancer Res 2017;29:436–70. [PubMed] DOI:10.21147/j.issn.1000-9604.2017.05.11
- Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563–7. [PubMed] DOI:10.1038/nature14011
- Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000;192:1027–34. [PubMed] DOI:10.1084/jem.192.7.1027
- Dong H, Zhu G, Tamada K, et al. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med 1999;5:1365–9. [PubMed] DOI:10.1038/70932
- Butte MJ, Keir ME, Phamduy TB, et al. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 2007;27:111–22. [PubMed] DOI:10.1016/j.immuni.2007.05.016
- Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis 2015;6:e1792. [PubMed] DOI:10.1038/cddis.2015.162
- Blackburn SD, Shin H, Haining WN, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection . Nat Immunol 2009;10:29–37. [PubMed] DOI:10.1038/ni.1679
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med 2016;8:328rv4. [PubMed] DOI:10.1126/scitranslmed.aad7118
- Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015;161:205–14. [PubMed] DOI:10.1016/j.cell.2015.03.030
- Sharma P, Hu-Lieskovan S, Wargo JA, et al. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017;168:707–23. [PubMed] DOI:10.1016/j.cell.2017.01.017
- Ishida Y, Agata Y, Shibahara K, et al. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992;11:3887–95. [PubMed]
- Nishimura H, Okazaki T, Tanaka Y, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001;291:319–22. [PubMed] DOI:10.1126/science.291.5502.319
- Dong H, Zhu G, Tamada K, et al. B7-H1 determines accumulation and deletion of intrahepatic CD8+ T lymphocytes . Immunity 2004;20:327–36. [PubMed] DOI:10.1016/S1074-7613(04)00050-0
- Park JJ, Omiya R, Matsumura Y, et al. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood 2010;116:1291–8. [PubMed] DOI:10.1182/blood-2010-01-265975
- Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2001;2:261–8. [PubMed] DOI:10.1038/85330
- Xiao Y, Yu S, Zhu B, et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J Exp Med 2014;211:943–59. [PubMed] DOI:10.1084/jem.20130790
- Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002;8:793–800. [PubMed] DOI:10.1038/nm730
- Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017;545:495–9. [PubMed] DOI:10.1038/nature22396
- Hirano F, Kaneko K, Tamura H, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res 2005;65:1089–96. [PubMed]
- Marzec M, Zhang Q, Goradia A, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A 2008;105:20852–7. [PubMed] DOI:10.1073/pnas.0810958105
- Noman MZ, Desantis G, Janji B, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 2014;211:781–90. [PubMed] DOI:10.1084/jem.20131916
- Lim TS, Chew V, Sieow JL, et al. PD-1 expression on dendritic cells suppresses CD8+ T cell function and antitumor immunity. Oncoimmunology 2015;5:e1085146. [PubMed] DOI:10.1080/2162402X.2015.1085146
- Ni L, Ma CJ, Zhang Y, et al. PD-1 modulates regulatory T cells and suppresses T-cell responses in HCV-associated lymphoma. Immunol Cell Biol 2011;89:535–9. [PubMed] DOI:10.1038/icb.2010.121
- Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol 2008;8:467–77. [PubMed] DOI:10.1038/nri2326
- Peng W, Liu C, Xu C, et al. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-γ inducible chemokines. Cancer Res 2012;72:5209–18. [PubMed] DOI:10.1158/0008-5472.CAN-12-1187
- Wong RM, Scotland RR, Lau RL, et al. Programmed death-1 blockade enhances expansion and functional capacity of human melanoma antigen-specific CTLs. Int Immunol 2007;19:1223–34. [PubMed] DOI:10.1093/intimm/dxm091
- Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65. [PubMed] DOI:10.1056/NEJMoa1200694
- Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 2016;7:10501. [PubMed] DOI:10.1038/ncomms10501
- Pauken KE, Sammons MA, Odorizzi PM, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016;354:1160–5. [PubMed] DOI:10.1126/science.aaf2807
- Ngiow SF, Young A, Jacquelot N, et al. A threshold level of intratumor CD8+ T-cell PD1 expression dictates therapeutic response to anti-PD1. Cancer Res 2015;75:3800–11. [PubMed] DOI:10.1158/0008-5472.CAN-15-1082
- Fisher DT, Chen Q, Appenheimer MM, et al. Hurdles to lymphocyte trafficking in the tumor microenvironment: implications for effective immunotherapy. Immunol Invest 2006;35:251–77. [PubMed] DOI:10.1080/08820130600745430
- Liu H, Shen J, Lu K. IL-6 and PD-L1 blockade combination inhibits hepatocellular carcinoma cancer development in mouse model. Biochem Biophys Res Commun 2017;486:239–44. [PubMed] DOI:10.1016/j.bbrc.2017.02.128
- Sun Z, Fourcade J, Pagliano O, et al. IL10 and PD-1 cooperate to limit the activity of tumor-specific CD8+ T Cells. Cancer Res 2015;75:1635–44. [PubMed] DOI:10.1158/0008-5472.CAN-14-3016
- Terme M, Pernot S, Marcheteau E, et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res 2013;73:539–49. [PubMed] DOI:10.1158/0008-5472.CAN-12-2325
- Shin DS, Zaretsky JM, Escuin-Ordinas H, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov 2017;7:188–201. [PubMed] DOI:10.1158/2159-8290.CD-16-1223
- Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 2012;4:127ra37. [PubMed] DOI:10.1126/scitranslmed.3003689
- Haworth KB, Leddon JL, Chen CY, et al. Going back to class I: MHC and immunotherapies for childhood cancer. Pediatr Blood Cancer 2015;62:571–6. [PubMed] DOI:10.1002/pbc.25359
- Holmgaard RB, Zamarin D, Munn DH, et al. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med 2013;210:1389–402. [PubMed] DOI:10.1084/jem.20130066
- Allard B, Pommey S, Smyth MJ, et al. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res 2013;19:5626–35. [PubMed] DOI:10.1158/1078-0432.CCR-13-0545
- Arlauckas SP, Garris CS, Kohler RH, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy . Sci Transl Med 2017;9. [PubMed] DOI:10.1126/scitranslmed.aal3604
- Li JY, Duan XF, Wang LP, et al. Selective depletion of regulatory T cell subsets by docetaxel treatment in patients with nonsmall cell lung cancer. J Immunol Res 2014;2014:286170. [PubMed] DOI:10.1155/2014/286170
- Peng J, Hamanishi J, Matsumura N, et al. Chemotherapy induces programmed cell death-ligand 1 overexpression via the nuclear factor-κB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res 2015;75:5034–45. [PubMed] DOI:10.1158/0008-5472.CAN-14-3098
- Black M, Barsoum IB, Truesdell P, et al. Activation of the PD-1/PD-L1 immune checkpoint confers tumor cell chemoresistance associated with increased metastasis. Oncotarget 2016;7:10557–67. [PubMed] DOI:10.18632/oncotarget.7235
- Mkrtichyan M, Najjar YG, Raulfs EC, et al. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Eur J Immunol 2011;41:2977–86. [PubMed] DOI:10.1002/eji.201141639
- Sharma P, Retz M, Siefker-Radtke A, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol 2017;18:312–22. [PubMed] DOI:10.1016/S1470-2045(17)30065-7
- Shahda S, Noonan AM, Bekaii-Saab TS, et al. A phase II study of pembrolizumab in combination with mFOLFOX6 for patients with advanced colorectal cancer. J Clin Oncol 2017;35(Suppl):abstract 3541.
- Fuchs CS, DoiT, Jang R, et al. KEYNOTE-059 cohort 1: Efficacy and safety of pembrolizumab (pembro) monotherapy in patients with previously treated advanced gastric cancer. J Clin Oncol 2017;35(Suppl):abstract 4003.
- Burnette BC, Liang H, Lee Y, et al. The efficacy of radiotherapy relies upon induction of type I interferon-dependent innate and adaptive immunity. Cancer Res 2011;71:2488–96. [PubMed] DOI:10.1158/0008-5472.CAN-10-2820
- Demaria S, Ng B, Devitt ML, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys 2004;58:862–70. [PubMed] DOI:10.1016/j.ijrobp.2003.09.012
- Deng L, Liang H, Burnette B, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 2014;124:687–95. [PubMed] DOI:10.1172/JCI67313
- Zeng J, See AP, Phallen J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys 2013;86:343–9. [PubMed] DOI:10.1016/j.ijrobp.2012.12.025
- Haymaker CL, Kim D, Uemura M, et al. Metastatic melanoma patient had a complete response with clonal expansion after whole brain radiation and PD-1 blockade. Cancer Immunol Res 2017;5:100–5. [PubMed] DOI:10.1158/2326-6066.CIR-16-0223
- Aguilera TA, Giaccia AJ. Molecular pathways: oncologic pathways and their role in T-cell exclusion and immune evasion — A new role for the AXL receptor tyrosine kinase. Clin Cancer Res 2017;23:2928–33. [PubMed] DOI:10.1158/1078-0432.CCR-17-0189
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62–8. [PubMed] DOI:10.1126/science.aaa4967
- Bonini C, Mondino A. Adoptive T-cell therapy for cancer: The era of engineered T cells. Eur J Immunol 2015;45:2457–69. [PubMed] DOI:10.1002/eji.201545552
- Moon EK, Ranganathan R, Eruslanov E, et al. Blockade of programmed death 1 augments the ability of human T cells engineered to target NY-ESO-1 to control tumor growth after adoptive transfer. Clin Cancer Res 2016;22:436–47. [PubMed] DOI:10.1158/1078-0432.CCR-15-1070
- Cherkassky L, Morello A, Villena-Vargas J, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest 2016;126:3130–44. [PubMed] DOI:10.1172/JCI83092
- Schneider H, Downey J, Smith A, et al. Reversal of the TCR stop signal by CTLA-4. Science 2006;313:1972–5. [PubMed] DOI:10.1126/science.1131078
- Liu J, Zhang S, Hu Y, et al. Targeting PD-1 and Tim-3 pathways to reverse CD8 T-Cell exhaustion and enhance ex vivo T-Cell responses to autologous dendritic/tumor vaccines. J Immunother 2016;39:171–80. [PubMed] DOI:10.1097/CJI.0000000000000122
- Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A 2010;107:7875–80. [PubMed] DOI:10.1073/pnas.1003345107
- Chauvin JM, Pagliano O, Fourcade J, et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients . J Clin Invest 2015;125:2046–58. [PubMed] DOI:10.1172/JCI80445
- Wing K, Onishi Y, Prieto-Martin P, et al. CTLA-4 control over Foxp3+ regulatory T cell function . Science 2008;322:271–5. [PubMed] DOI:10.1126/science.1160062
- Kvistborg P, Philips D, Kelderman S, et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response . Sci Transl Med 2014;6:254ra128. [PubMed] DOI:10.1126/scitranslmed.3008918
- Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372:2006–17. [PubMed] DOI:10.1056/NEJMoa1414428
- Kim JE, Patel MA, Mangraviti A, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res 2017;23:124–36. [PubMed] DOI:10.1158/1078-0432.CCR-15-1535
- Fourcade J, Sun Z, Pagliano O, et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8+ T cells induced by melanoma vaccines . Cancer Res 2014;74:1045–55. [PubMed] DOI:10.1158/0008-5472.CAN-13-2908
- Dougall WC, Kurtulus S, Smyth MJ, et al. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev 2017;276:112–20. [PubMed] DOI:10.1111/imr.12518
- Schlaphoff V, Lunemann S, Suneetha PV, et al. Dual function of the NK cell receptor 2B4 (CD244) in the regulation of HCV-specific CD8+ T cells. PLoS Pathog 2011;7:e1002045. [PubMed] DOI:10.1371/journal.ppat.1002045
- Kared H, Saeed S, Klein MB, et al. CD127 expression, exhaustion status and antigen specific proliferation predict sustained virologic response to IFN in HCV/HIV co-infected individuals. PLoS One 2014;9:e101441. [PubMed] DOI:10.1371/journal.pone.0101441
- Fourcade J, Sun Z, Pagliano O, et al. CD8+ T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1 . Cancer Res 2012;72:887–96. [PubMed] DOI:10.1158/0008-5472.CAN-11-2637
- Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol 2009;9:271–85. [PubMed] DOI:10.1038/nri2526
- Gros A, Robbins PF, Yao X, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors . J Clin Invest 2014;124:2246–59. [PubMed] DOI:10.1172/JCI73639
- Mardiana S, John LB, Henderson MA, et al. A multifunctional role for adjuvant anti-4-1BB therapy in augmenting antitumor response by chimeric antigen receptor T cells. Cancer Res 2017;77:1296–309. [PubMed] DOI:10.1158/0008-5472.CAN-16-1831
- Chen S, Lee LF, Fisher TS, et al. Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol Res 2015;3:149–60. [PubMed] DOI:10.1158/2326-6066.CIR-14-0118
- Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res 2013;73:6900–12. [PubMed] DOI:10.1158/0008-5472.CAN-13-1550
- Guo Z, Wang X, Cheng D, et al. PD-1 blockade and OX40 triggering synergistically protects against tumor growth in a murine model of ovarian cancer. PLoS One 2014;9:e89350. [PubMed] DOI:10.1371/journal.pone.0089350
- Buchan S, Manzo T, Flutter B, et al. OX40- and CD27-mediated costimulation synergizes with anti-PD-L1 blockade by forcing exhausted CD8+ T cells to exit quiescence . J Immunol 2015;194:125–33. [PubMed] DOI:10.4049/jimmunol.1401644
- Isogawa M, Chung J, Murata Y, et al. CD40 activation rescues antiviral CD8+ T cells from PD-1-mediated exhaustion . PLoS Pathog 2013;9:e1003490. [PubMed] DOI:10.1371/journal.ppat.1003490
- Winograd R, Byrne KT, Evans RA, et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res 2015;3:399–411. [PubMed] DOI:10.1158/2326-6066.CIR-14-0215
- Ebert PJR, Cheung J, Yang Y, et al. MAP Kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 2016;44:609–21. [PubMed] DOI:10.1016/j.immuni.2016.01.024
- Bengsch B, Johnson AL, Kurachi M, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion . Immunity 2016;45:358–73. [PubMed] DOI:10.1016/j.immuni.2016.07.008
- Shalapour S, Karin M. Fatty acid-induced T cell loss greases liver carcinogenesis. Cell Metab 2016;23:759–61. [PubMed] DOI:10.1016/j.cmet.2016.04.018
- Patsoukis N, Bardhan K, Chatterjee P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun 2015;6:6692. [PubMed] DOI:10.1038/ncomms7692
- Yang W, Bai Y, Xiong Y, et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism . Nature 2016;531:651–5. [PubMed] DOI:10.1038/nature17412
- Chen X, Song M, Zhang B, et al. Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxid Med Cell Longev 2016;2016:1580967. [PubMed] DOI:10.1155/2016/1580967
- Doedens AL, Phan AT, Stradner MH, et al. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen . Nat Immunol 2013;14:1173–82. [PubMed] DOI:10.1038/ni.2714
- Peng D, Kryczek I, Nagarsheth N, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015;527:249–53. [PubMed] DOI:10.1038/nature15520
- Clancy-Thompson E, Perekslis TJ, Croteau W, et al. Melanoma induces, and adenosine suppresses, CXCR3-cognate chemokine production and T-cell infiltration of lungs bearing metastatic-like disease. Cancer Immunol Res 2015;3:956–67. [PubMed] DOI:10.1158/2326-6066.CIR-15-0015
- Flecken T, Schmidt N, Hild S, et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma . Hepatology 2014;59:1415–26. [PubMed] DOI:10.1002/hep.26731
- Chen Y, Ramjiawan RR, Reiberger T, et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology 2015;61:1591–602. [PubMed] DOI:10.1002/hep.27665
- Sato T, McCue P, Masuoka K, et al. Interleukin 10 production by human melanoma. Clin Cancer Res 1996;2:1383–90. [PubMed]
- Lee KA, Shin KS, Kim GY, et al. Characterization of age-associated exhausted CD8+ T cells defined by increased expression of Tim-3 and PD-1 . Aging Cell 2016;15:291–300. [PubMed] DOI:10.1111/acel.12435
- Nguyen DP, Li J, Tewari AK. Inflammation and prostate cancer: the role of interleukin 6 (IL-6). BJU Int 2014;113:986–92. [PubMed] DOI:10.1111/bju.12452
- Thomas DA, Massagué J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005;8:369–80. [PubMed] DOI:10.1016/j.ccr.2005.10.012
- Park BV, Freeman ZT, Ghasemzadeh A, et al. TGFβ1-mediated SMAD3 enhances PD-1 expression on antigen-specific T cells in cancer. Cancer Discov 2016;6:1366–81. [PubMed] DOI:10.1158/2159-8290.CD-15-1347
- Young MR, Wright MA, Coogan M, et al. Tumor-derived cytokines induce bone marrow suppressor cells that mediate immunosuppression through transforming growth factor beta. Cancer Immunol Immunother 1992;35:14–8. [PubMed]
- Baas M, Besançon A, Goncalves T, et al. TGFβ-dependent expression of PD-1 and PD-L1 controls CD8+ T cell anergy in transplant tolerance . Elife 2016;5:e08133. [PubMed] DOI:10.7554/eLife.08133
- Choueiri TK, Figueroa DJ, Fay AP, et al. Correlation of PD-L1 tumor expression and treatment outcomes in patients with renal cell carcinoma receiving sunitinib or pazopanib: results from COMPARZ, a randomized controlled trial. Clin Cancer Res 2015;21:1071–7. [PubMed] DOI:10.1158/1078-0432.CCR-14-1993
- Voron T, Colussi O, Marcheteau E, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors . J Exp Med 2015;212:139–48. [PubMed] DOI:10.1084/jem.20140559
- Kaneda MM, Messer KS, Ralainirina N, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016;539:437–42. [PubMed] DOI:10.1038/nature19834
- De Henau O, Rausch M, Winkler D, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 2016;539:443–7. [PubMed] DOI:10.1038/nature20554
- Antonioli L, Blandizzi C, Pacher P, et al. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer 2013;13:842–57. [PubMed] DOI:10.1038/nrc3613
- Beavis PA, Stagg J, Darcy PK, et al. CD73: a potent suppressor of antitumor immune responses. Trends Immunol 2012;33:231–7. [PubMed] DOI:10.1016/j.it.2012.02.009
- Nakanishi M, Rosenberg DW. Multifaceted roles of PGE2 in inflammation and cancer. Semin Immunopathol 2013;35:123–37. [PubMed] DOI:10.1007/s00281-012-0342-8
- Zelenay S, van der Veen AG, Böttcher JP, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 2015;162:1257–70. [PubMed] DOI:10.1016/j.cell.2015.08.015
- Chen JH, Perry CJ, Tsui YC, et al. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat Med 2015;21:327–34. [PubMed] DOI:10.1038/nm.3831
- Wainwright DA, Chang AL, Dey M, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res 2014;20:5290–301. [PubMed] DOI:10.1158/1078-0432.CCR-14-0514
- Sai J, Owens P, Novitskiy SV, et al. PI3K inhibition reduces mammary tumor growth and facilitates antitumor immunity and anti-PD1 responses. Clin Cancer Res 2017;23:3371–84. [PubMed] DOI:10.1158/1078-0432.CCR-16-2142
- Peng W, Chen JQ, Liu C, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov 2016;6:202–16. [PubMed] DOI:10.1158/2159-8290.CD-15-0283
- Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015;523:231–5. [PubMed] DOI:10.1038/nature14404
- Akbay EA, Koyama S, Carretero J, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov 2013;3:1355–63. [PubMed] DOI:10.1158/2159-8290.CD-13-0310
- Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124–8. [PubMed] DOI:10.1126/science.aaa1348
- Van Allen EM, Miao D, Schilling B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350:207–11. [PubMed] DOI:10.1126/science.aad0095
- Le DT, Uram JN, Wang H, et al. PD-1 blockade in mismatch repair deficient non-colorectal cancers. J Clin Oncol 2016;34(Suppl):195. DOI:10.1200/jco.2016.34.4_suppl.195>
- Stevanovic S, Pasetto A, Helman SR, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 2017;356:200–5. [PubMed] DOI:10.1126/science.aak9510
- Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 2015;372:311–9. [PubMed] DOI:10.1056/NEJMoa1411087
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30. [PubMed] DOI:10.1056/NEJMoa1412082
- Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med 2016;375:1823–33. [PubMed] DOI:10.1056/NEJMoa1606774
- Berger R, Rotem-Yehudar R, Slama G, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res 2008;14:3044–51. [PubMed] DOI:10.1158/1078-0432.CCR-07-4079
- Armand P, Nagler A, Weller EA, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol 2013;31:4199–206. [PubMed] DOI:10.1200/JCO.2012.48.3685
- Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014;515:558–62. [PubMed] DOI:10.1038/nature13904
- McDermott DF, Sosman JA, Sznol M, et al. Atezolizumab, an anti-programmed death-ligand 1 antibody, in metastatic renal cell carcinoma: long-term safety, clinical activity, and immune correlates from a phase Ia study. J Clin Oncol 2016;34:833–42. [PubMed] DOI:10.1200/JCO.2015.63.7421
- Balar AV, Galsky MD, Rosenberg JE, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet 2017;389:67–76. [PubMed] DOI:10.1016/S0140-6736(16)32455-2
- Hodi FS, Chesney J, Pavlick AC, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol 2016;17:1558–68. [PubMed] DOI:10.1016/S1470-2045(16)30366-7
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34. [PubMed] DOI:10.1056/NEJMoa1504030
- Kanda S, Goto K, Shiraishi H, et al. Safety and efficacy of nivolumab and standard chemotherapy drug combination in patients with advanced non-small-cell lung cancer: a four arms phase Ib study. Ann Oncol 2016;27:2242–50. [PubMed] DOI:10.1093/annonc/mdw416
- Liniker E, Menzies AM, Kong BY, et al. Activity and safety of radiotherapy with anti-PD-1 drug therapy in patients with metastatic melanoma. Oncoimmunology 2016;5:e1214788. [PubMed] DOI:10.1080/2162402X.2016.1214788