
Novel circular RNA expression profile of uveal melanoma revealed by microarray
Introduction
Uveal melanoma (UM) is the most common primary intraocular malignancy in adults, and it seriously threatens vision, the eyeballs and even the lives of patients (1). UM arises from melanocytes in the uveal tract, the middle vascular layer of the eyeball wall, residing in the iris, ciliary body, or choroid. UM is characterized by a poor prognosis and early metastasis. Current treatment methods for UM include brachytherapy, proton beam irradiation, transpupillary thermotherapy, local resection, and enucleation (2,3). However, regardless of the existing treatments used for tumors in the eye, approximately half of the patients die from tumor metastasis. This metastasis may occur early in the disease and often spreads to the liver (4). Metastatic UM patients have a high mortality rate and a dismal prognosis. Presently, no significantly effective local treatment is available for UM, and systemic treatment lacks effective drugs and interventions. Despite the development of relevant studies, the 5-year survival rate of UM patients has not changed in the past 30 years (5,6). UM patients face treatment difficulties, serious visual functional impairment, and high mortality (7). Therefore, to develop novel and effective therapeutic approaches, understanding the molecular mechanism of UM development and progression is important.
Circular RNA (circRNA) is a new type of endogenous noncoding RNA recognized as a stable class of regulatory transcripts in mammals (8-10). circRNAs have a characteristic covalently closed loop structure, which differs from linear RNA terminated with 5’ caps and 3’ tails, and they do not have 5’ to 3’ polarity or polyadenylated tails (11,12). circRNAs, produced by a “back-splice” reaction (13,14), are abundant, conserved and stable across species and are specifically expressed in specific cell types or stages of development (15). Previously considered a transcriptional error or by-product, circRNA has recently been shown to play an important role in many physiological and pathophysiological processes (16,17). Specifically, because circRNA is resistant to the degradation of RNase R or RNA exonuclease (18) and contains selectively conserved microRNA (miRNA) target sites, circRNA acts as an effective miRNA sponge and interacts with miRNAs to regulate gene expression (19,20). miRNAs are small, non-coding single strand RNA molecules that play important roles in RNA silencing and post-transcriptional regulation of gene expression (21). Many research groups have examined the dysregulation of miRNAs in UM and how miRNAs affect tumor growth and metastasis (22,23), exerting potential roles as circulating markers of UM. In addition, circRNA has many functions, such as the regulation of alternative splicing and expression of transcription or parental genes, with some studies confirming that endogenous circRNA encodes a functional protein in human cells (24-27). In recent years, a growing number of studies has suggested that circRNA shows strong functional potential in regulating apoptosis, regulating proliferation, angiogenesis and metastasis, indicating that circRNA can serve as a cancer biomarker and may be an important therapeutic target in cancer (28,29). However, studies regarding circRNA in UM have not been reported.
Because of the increasing importance of circRNAs in cancer, we speculate that circRNAs may be involved in the development of UM. To test our hypothesis, we first evaluated circRNA expression profiles in UM samples and normal uvea samples using microarray analysis. The dysregulated circRNAs in UM tissues were then verified by quantitative real-time polymerase chain reaction (qRT-PCR). A step-by-step bioinformatics analysis was performed to predict the pathways mediated by the validated circRNAs.
Materials and methods
Patients and tissue specimens
Fresh tumor tissues were obtained from 20 patients with UM at Beijing Tongren Hospital, China, from November 2016 to December 2017. All patients with UM underwent primary enucleation surgery. Twenty normal uvea tissues came from donors at the Beijing Tongren Eye Bank (Beijing, China). The normal uvea tissues were separated from the eyeballs within 18 h after death. All the patients had a pathological diagnosis of UM after surgery, and none was given radiation or chemotherapy therapy prior to the operation. The excised tissue samples were immediately stored in liquid nitrogen overnight after removal from the patients’ eyeball and then were transferred to −80 °C for storage until use. circRNA microarray analysis was performed using five UM samples and five normal controls. Twenty UM samples and 20 normal controls were used for validation by qRT-PCR. All subjects provided written informed consent before surgery, and all the study procedures were approved by the Institutional Review Board of Beijing Tongren Hospital.
RNA extraction
Total RNA containing small RNA was extracted from the tissue using the Trizol reagent (Invitrogen, Waltham, MA, USA), and the obtained RNA was purified using the mirVana miRNA Isolation Kit (Ambion, Austin, TX, USA). The purity and concentration of RNA were determined by spectrophotometer (NanoDrop ND-1000, Wilmington, USA) readings from OD260/280. The RNA integrity was determined by capillary electrophoresis using the RNA 6000 Nanochip Kit and Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). When the RNA extracts have RNA integrity values >6, further analysis can be performed.
circRNA microarray analysis
Total RNA extracted from tumor tissues (n=5) and normal uvea tissues (n=5) was used for microarray analysis. RNA digestion was performed using RNase R (Epicentre, Madison, WI, USA) to remove linear RNA. According to the manufacturer’s protocol of the CapitalBio cRNA Amplification and Labeling Kit (CapitalBio, Beijing, China), random primers were used to amplify the enriched circRNA and were subsequently transcribed into fluorescent circRNA. The amplified circRNA was purified using the RNeasy Mini kit (Qiagen, Düsseldorf, Germany). The labeled RNA was hybridized to the CapitalBiotech human circRNA Array V2.0. The circRNA array was designed to have four identical arrays per slide (4 × 180 K format), and each array contained probes to interrogate approximately 170,340 human circRNAs. These circRNA target sequences all came from Circbase, Deepbase and Rybak-Wolf 2015 (30). Each circRNA was detected simultaneously by one long probe and one short probe. The circRNA array also contained 4,974 Agilent control probes.
Microarray imaging and data analysis
circRNA microarray data were analyzed using GeneSpring Software Version 13.0 (Agilent, Santa Clara, USA) for data summary, standardization and quality control. To select genes that are differentially expressed, we used a fold change (FC) in expression ≥2.0 (P<0.05) of circRNAs between the UM and normal tissues samples for further analysis. The data were subjected to Log2 transformation using the adjusted data functions of CLUSTER 3.0 software and gene-centric, followed by hierarchical clustering using average linkage analysis. Finally, we used Java Treeview (Stanford University School of Medicine, Stanford, California, USA) for tree visualization.
qRT-PCR validation
Total RNA was extracted from 20 UM tissues and 20 normal uvea tissues using Trizol Reagent (Invitrogen), and the extracted RNA was reverse transcribed into cDNA using SuperScriptTM III Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions. First, 3 μg of RNA was mixed with Random N9 primer and dNTP Mix, and the mixture was placed on ice for 2 min and then was incubated at 65 °C for 5 min. The reverse transcription system was then prepared using the above mixture, RNase inhibitor and SuperScriptTM III Reverse Transcriptase. The reaction system was then incubated continuously for 1 min in water at 37 °C, incubated at 50 °C for 60 min, and then incubated at 70 °C for 15 min until reverse transcription was completed. qRT-PCR was performed in a ViiATM 7 real-time PCR system (Applied Biosystems, Wilmington, DE, USA) using qPCR SYBR Green master mix (CloudSeq Biotech Inc., Shanghai, China) and divergent primers designed for target circRNA [Sangon Biotech (Shanghai) Co., Ltd., China] to determine the expression of circRNAs. GAPDH [Sangon Biotech (Shanghai) Co., Ltd., China] was used as an internal control. Both the targeted circRNA and GAPDH were routinely amplified in triplicate with an annealing temperature of 60 °C. The relative expression levels of each target circRNA were calculated using the 2−ΔΔCt method. The primer sequences are presented in Table 1.

Full table
miRNA target gene prediction and enrichment analysis
circRNA plays an important role in miRNA function and transcription control as a competitive endogenous RNA (ceRNA) or positive regulator of its parental coding gene. A circRNA-miRNA network was constructed based on miRanda and TargetScan software to predict potential miRNA targets of differentially expressed circRNAs. Gene ontology (GO) analysis was performed to explore the functional role of target genes in biological processes, cellular components and molecular functions. The pathway analysis defined by the Kyoto Encyclopedia of Genes and Genomes (KEGG), BioCarta and Panther was determined by annotation, visualization and comprehensive discovery databases.
Statistical analysis
These works were created by GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). Student t-test was used to compare circRNA expression levels in tumor and normal uvea tissues and the results with the standard error of the mean. P<0.05 was considered statistically significant. IBM SPSS Statistics (Version 22.0; IBM Corp., New York, USA) software was used for data analysis.
Results
circRNA expression profiles in UM and normal uvea tissues
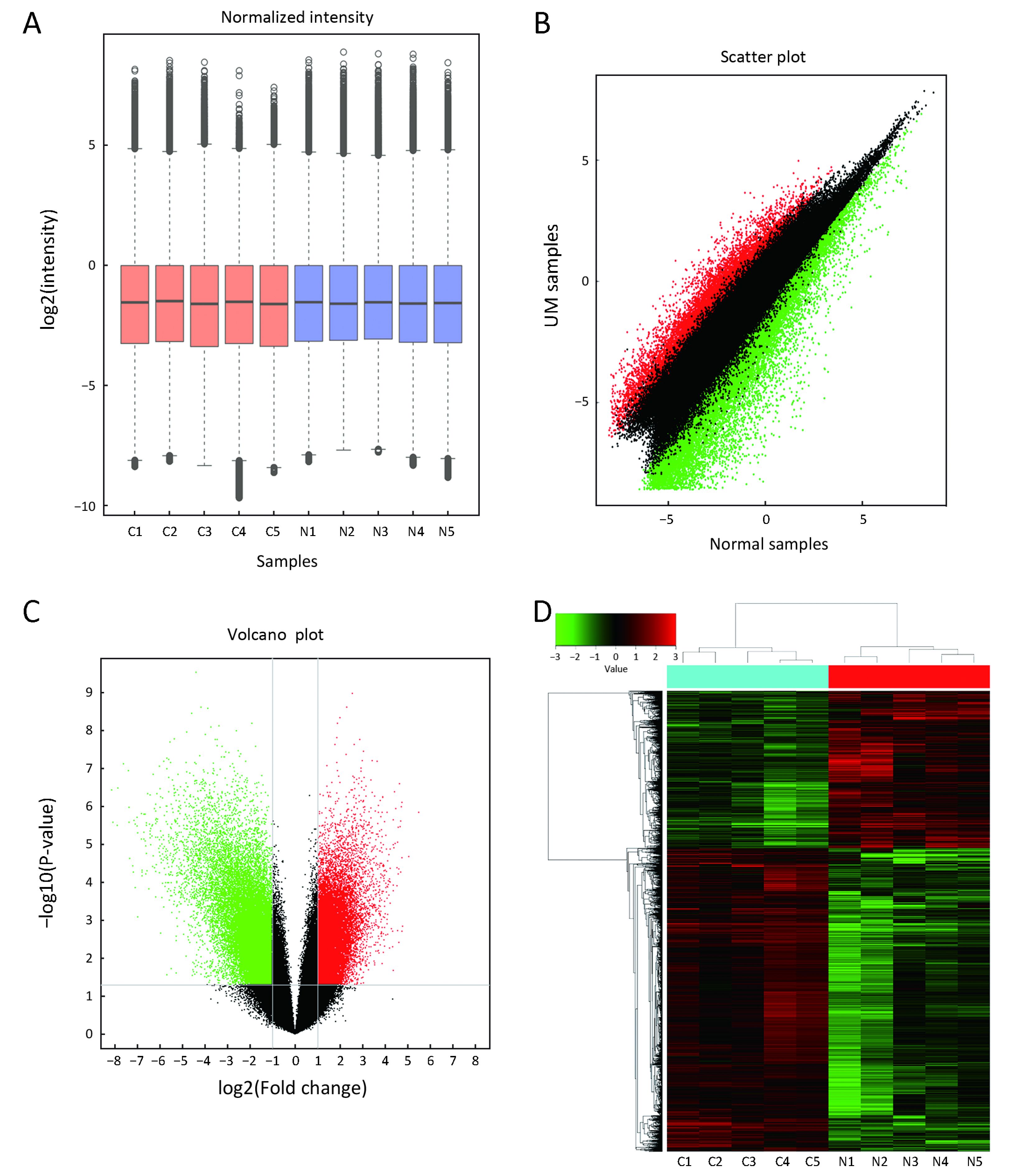
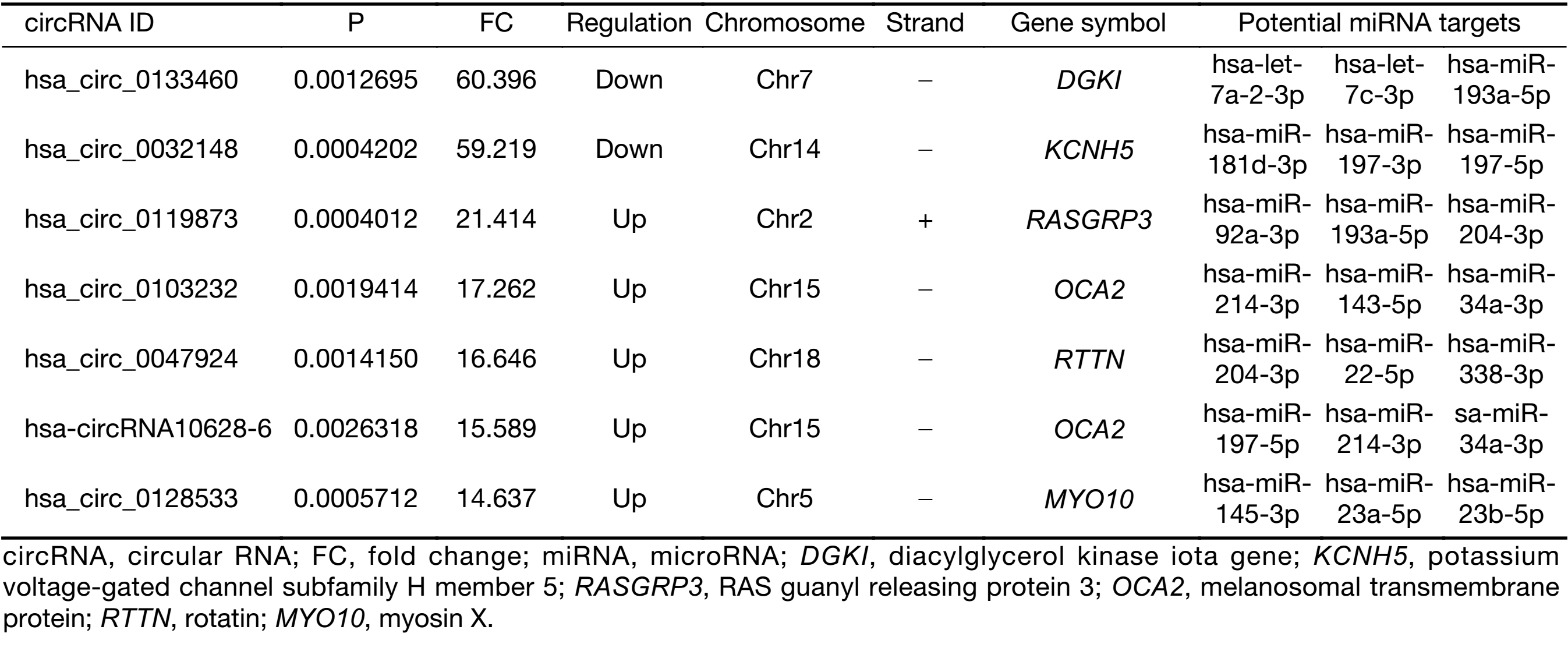
First, we characterized circRNA transcripts by circRNA microarray analysis of five UM samples and five normal uvea samples. In total, 170,340 circRNAs were detected in all samples. A box plot was drawn based on normalized signal values of the samples, which showed that the normalized intensity distributions of all datasets in the tumor and normal tissue samples are essentially similar (Figure 1A). Next, we screened the significantly aberrantly expressed circRNAs in UM using the statistical criteria for a FC≥2.0 and a P<0.05 between the UM and normal samples (Figure 1B). A volcano plot was drawn using the two factors of P value and FC value for the significantly differently expressed circRNAs obtained from the above analysis (Figure 1C). The hierarchical clustering analysis of the differentially expressed circRNAs clearly differentiated UM samples from normal uvea samples (Figure 1D). Therefore, 50,579 circRNAs were identified as being dysregulated in tumor tissues with FC≥2.0 and P<0.05. Among them, 20,654 were up-regulated and 29,925 were down-regulated in UM samples compared with normal uvea samples. The top 10 differentially expressed up-regulated and down-regulated circRNAs by FC value (absolute value) with potential functional targets are summarized in Table 2 and include the relevant FC values, P values, gene symbols, miRanda and TargetScan to predict the number of targeted miRNAs.
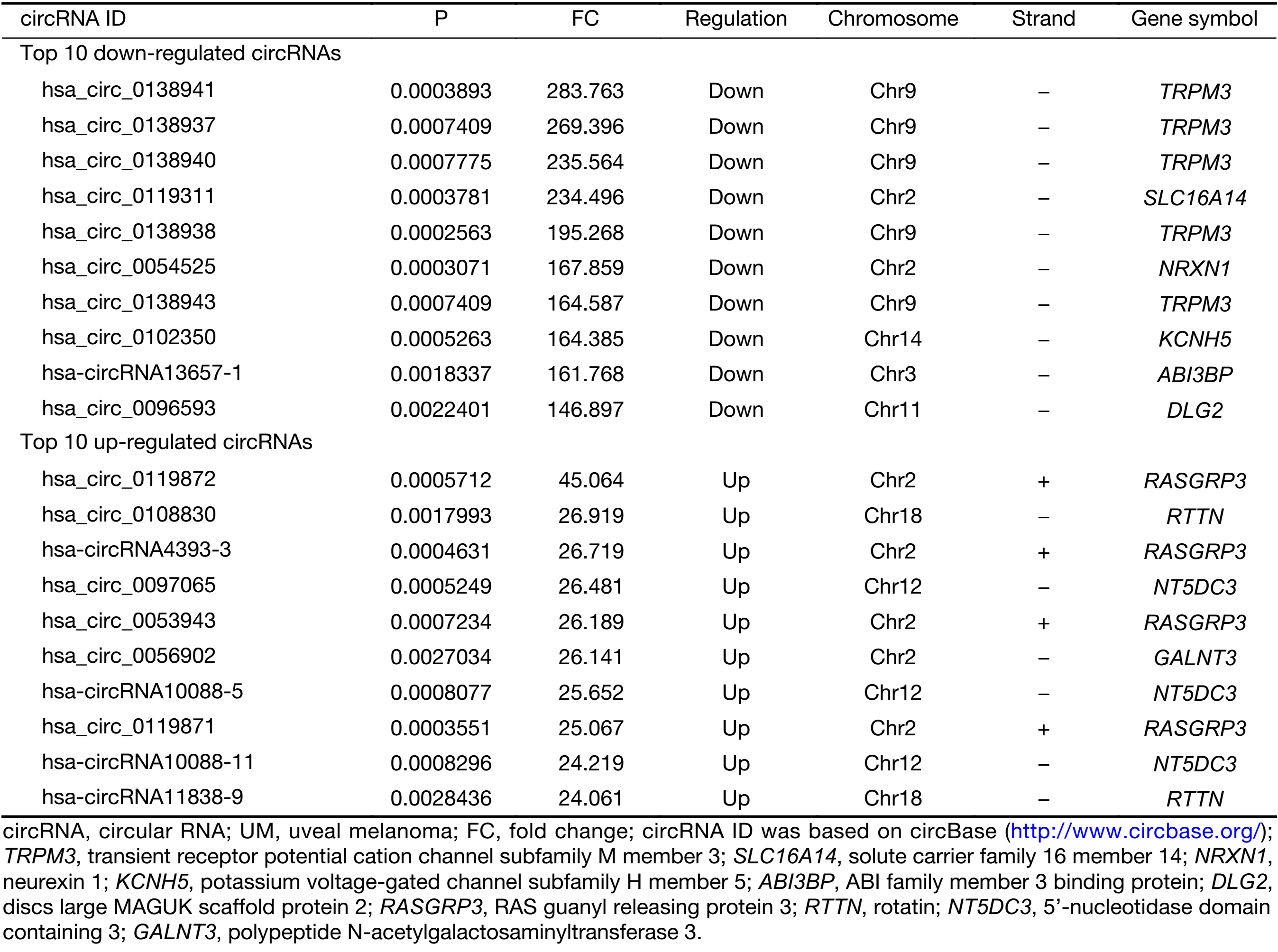
Full table
Validation of identified circRNAs by qRT-PCR
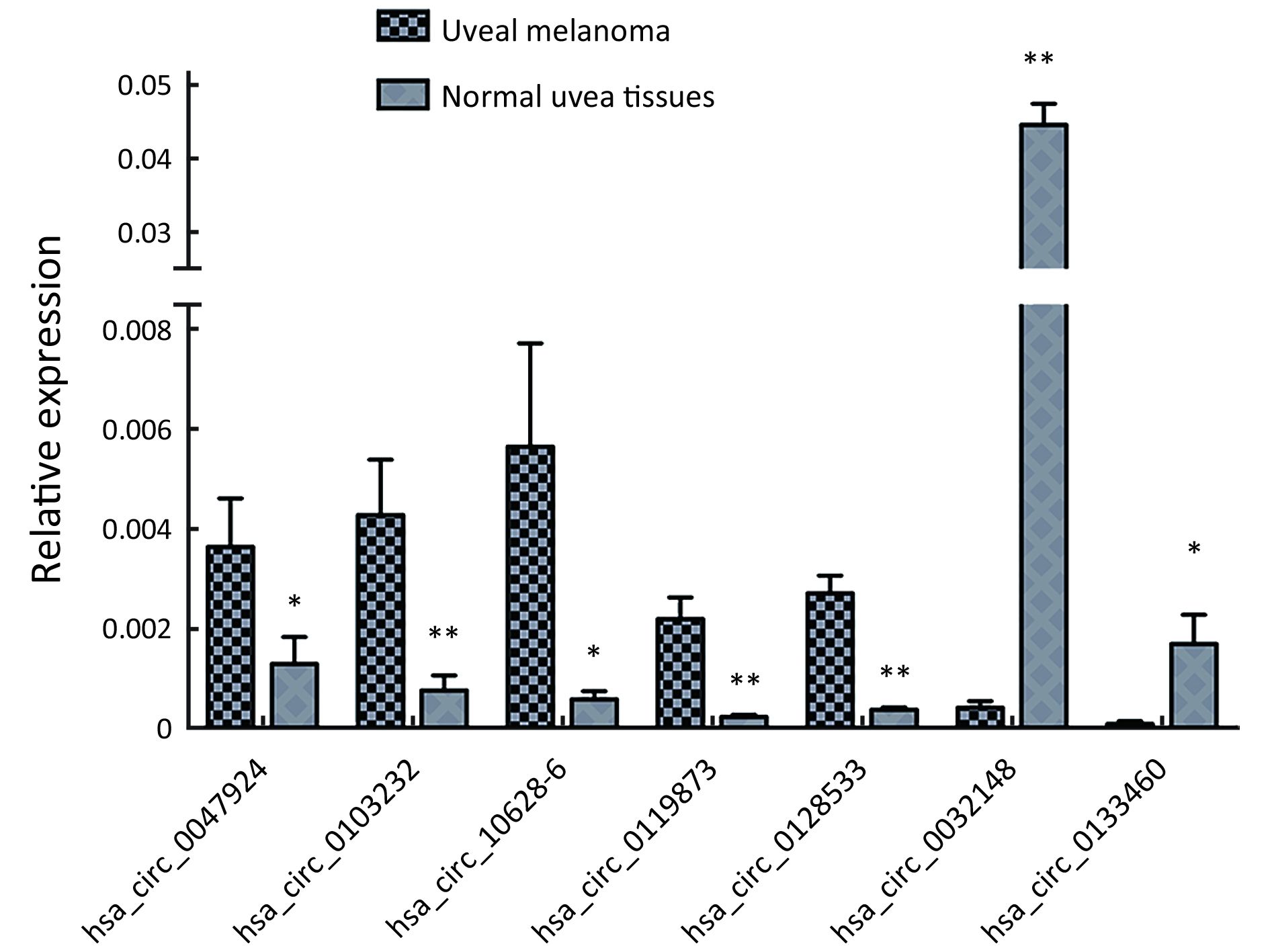
To validate the expression profiles of circRNAs in five UM tissues and five normal uvea tissues, we randomly selected 15 differentially expressed circRNAs, including 11 up-regulated circRNAs and four down-regulated circRNAs (Supplementary Table S1). We validated their expression levels using qRT-PCR in 20 pairs of UM tissues and normal uvea tissues. The divergent primers were designed to specifically amplify the backsplice sites of those circRNAs using gel electrophoresis and melt-curve analysis to confirm the validity of the PCR results. The expression levels of seven circRNAs, hsa_circ_0119873, hsa_circ_0128533, hsa_circ_0047924, hsa_circ_0103232 and hsa-circRNA10628-6, were significantly higher in uveal tumor tissues than in normal uvea tissues, and hsa_circ_0032148 and hsa_circ_0133460 were significantly lower in uveal tumor tissues than in normal uvea tissues. These results were consistent with the data of the microarray analysis in the scale-up validation test (Figure 2; *, P<0.05; **, P<0.01). These seven validated circRNAs were promising candidates for future analysis of molecular mechanisms involved in UM (Table 3).
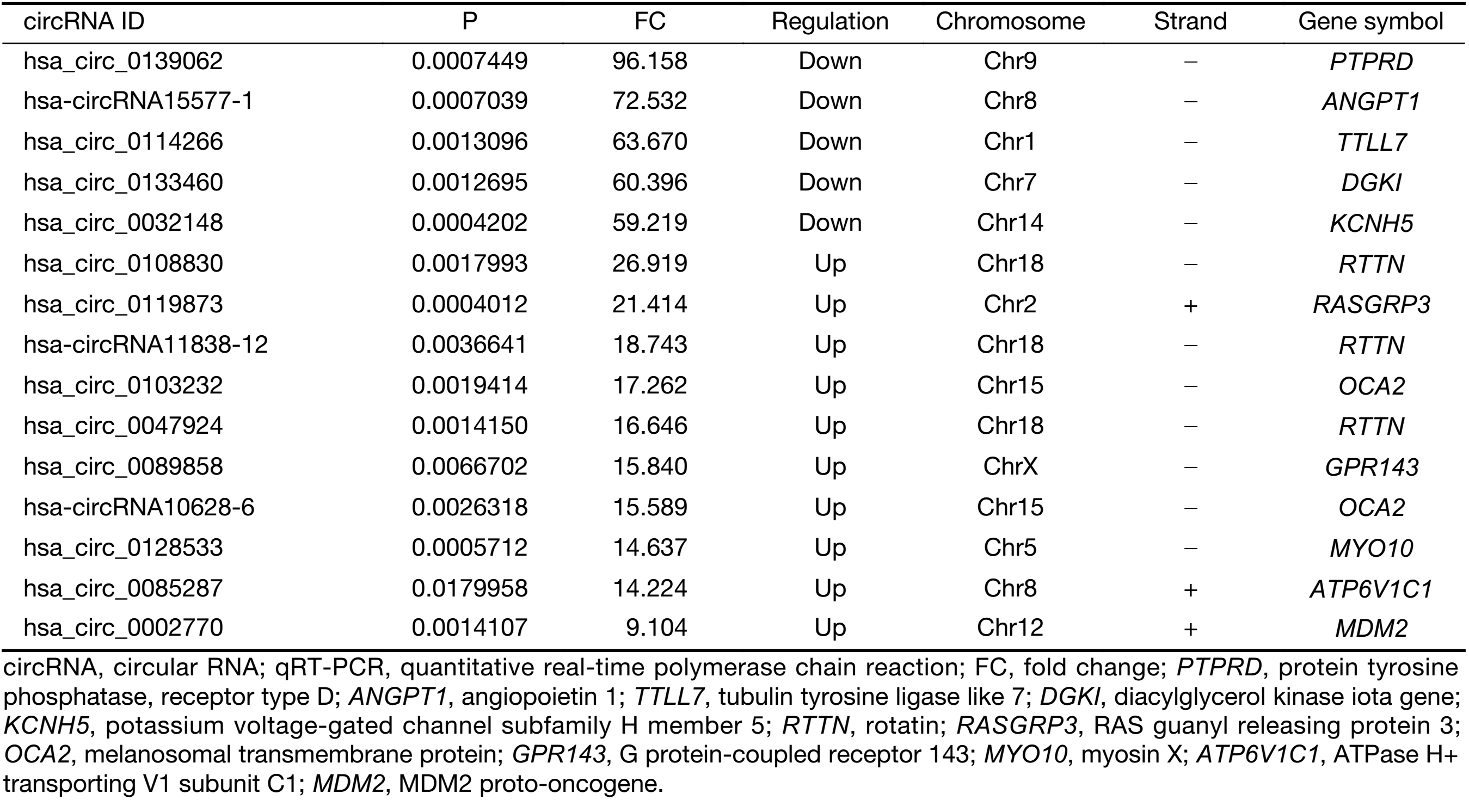
Full table
Full table
GO enrichment analysis and pathway analysis
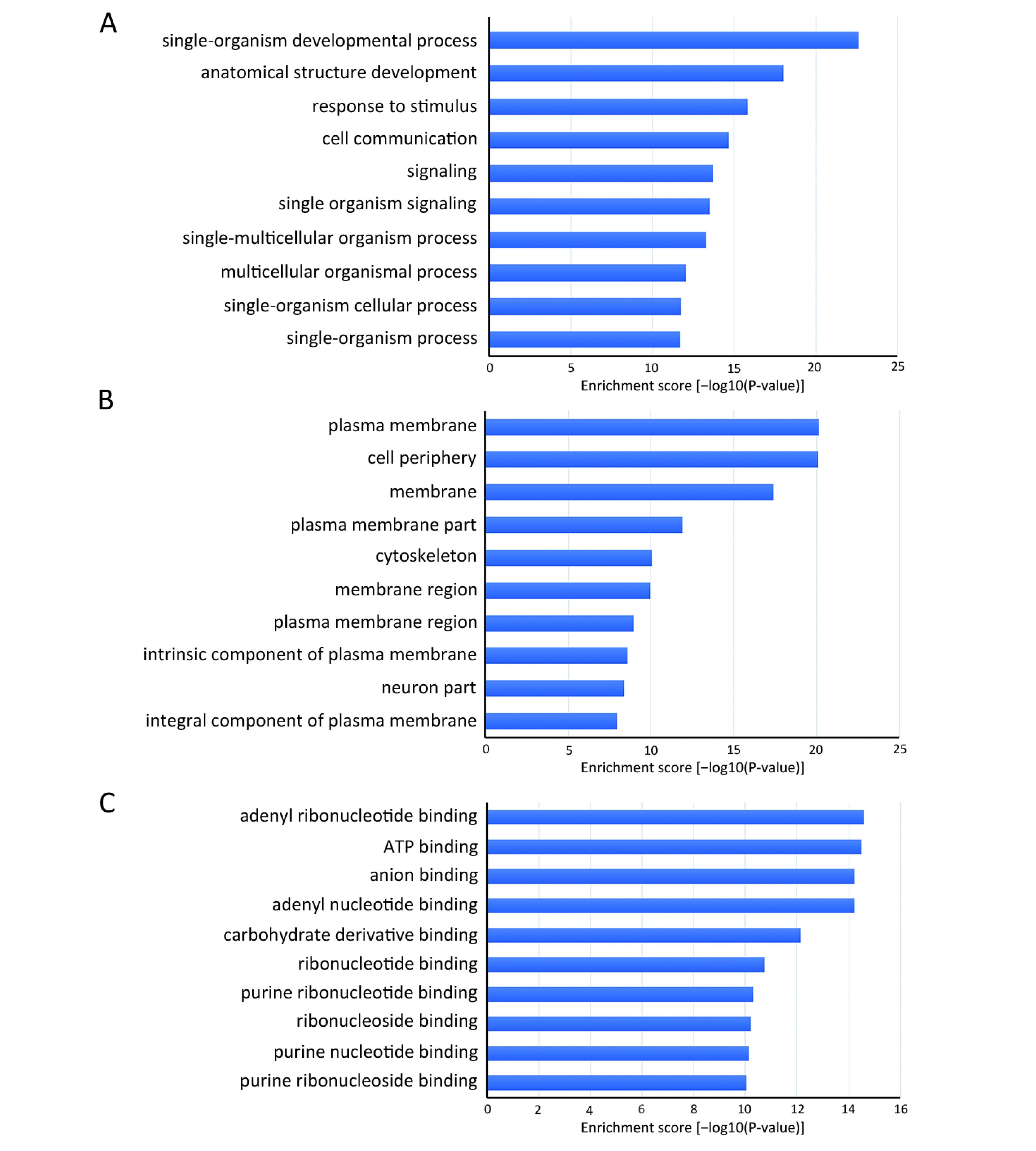
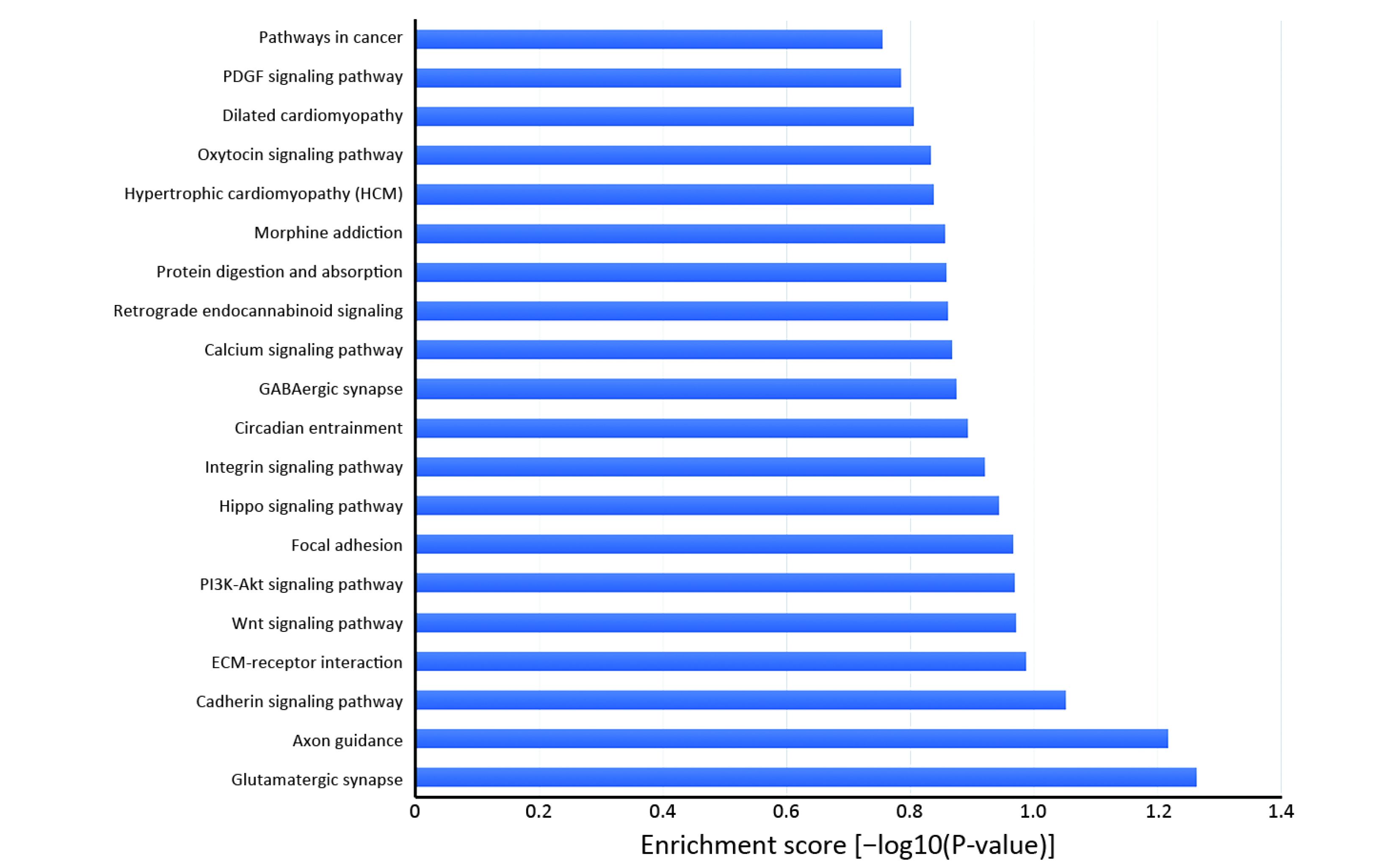
Many studies have confirmed that circRNAs play roles in multiple cellular processes of cancer by regulating gene expression at the transcriptional or post-transcriptional level. To explore the role of abnormally expressed circRNAs of UM tissues in biological processes, cellular components and molecular functions, we performed GO enrichment analysis of the gene symbols of those circRNAs to assess their properties (Figure 3). The lower the P value was, the more meaningful the enrichment was in GO analysis. The most significant biological process of GO enrichment analysis was single-organism developmental process (GO:0044767, P=2.05469e−12), the most significant cellular components of GO enrichment analysis was plasma membrane (GO:0005886, P=7.496023e−21), and the most significant major molecular function of GO enrichment analysis was adenyl ribonucleotide binding (GO:0032559, P=2.52808e−15). Furthermore, we performed pathway analysis of the genes using Panther, BioCarta and KEGG database. Twenty pathways, including the WNT signaling pathway, phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) signaling pathway and Hippo signaling pathway, were significantly enriched and are likely related to the roles of circRNAs in UM (Figure 4).
Prediction of miRNAs targeted by seven validated circRNAs
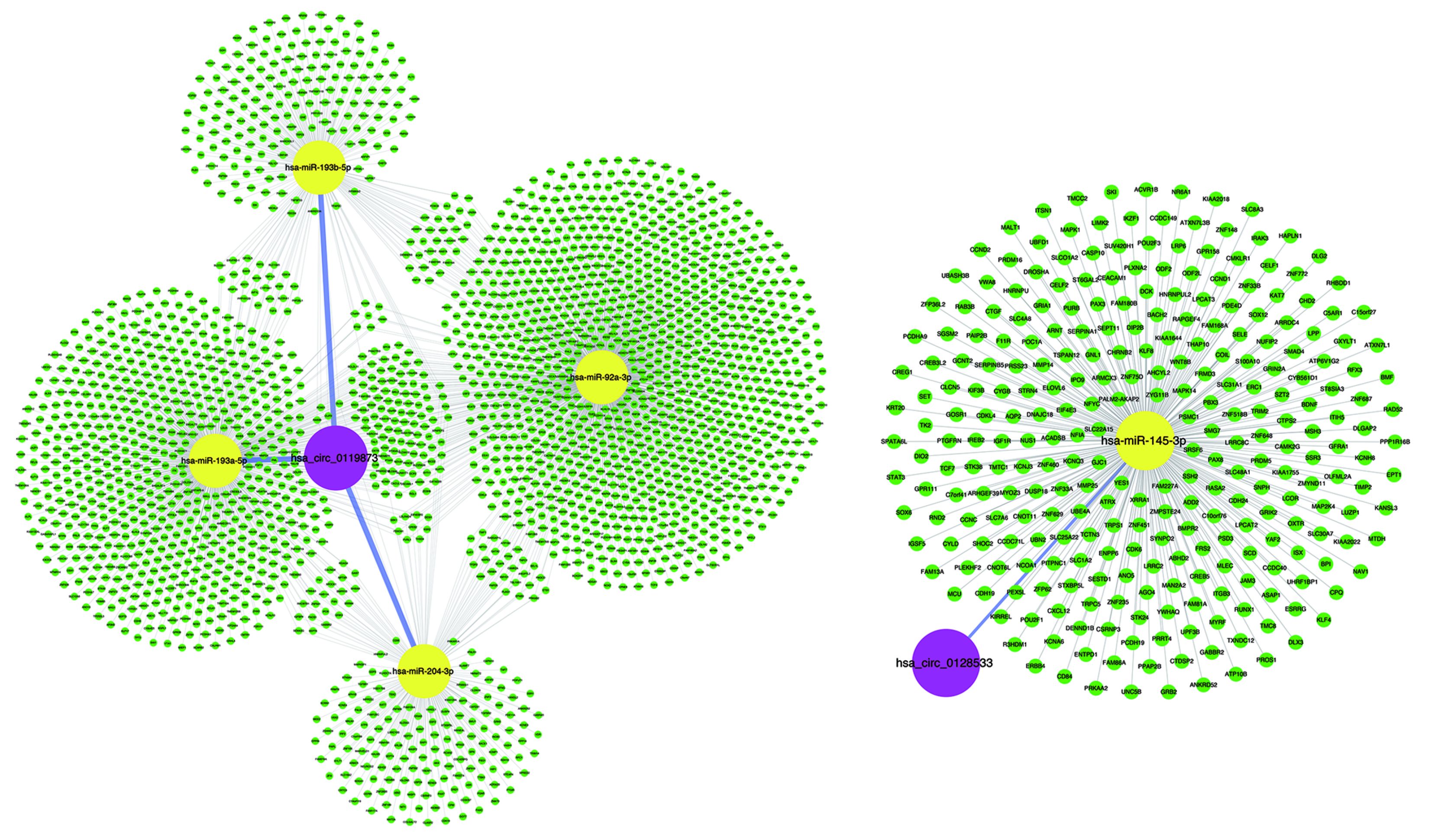
Recent studies have found that circRNA may function as a ceRNA that binds to miRNA (10,19). Therefore, differentially expressed circRNAs have functions as miRNA sponges by targeting miRNA response elements (MREs) (31). To explore circRNA/miRNA interactions in UM, we first determined that some of the miRNA targeted by the seven validated circRNAs were closely related to cancer from the published data. Next, we evaluated the potential function of circRNAs by

Discussion
UM is an extremely aggressive tumor, especially once metastasized, with high mortality. Many studies have found that this tumor invasion is closely related to the various molecular dysregulation processes of miRNAs (32), and it was reported that lncRNA abnormality in UM may be critical for the patient prognosis (33). circRNA is a circular noncoding RNA molecule that has been identified in recent years and has received new attention in the field of RNA. Increasing evidence has shown that various circRNAs are abnormally expressed in different cancer types and play important roles in many aspects of biology and disease, especially in cancer (34,35). For example, circMTO1 inhibits hepatocellular carcinoma progression by promoting p21 expression by acting as a sponge of oncogenic miR-9, demonstrating that circMTO1 is a potential target in hepatocellular carcinoma therapy (36). CircPVT1 promotes gastric cancer cell proliferation through a sponge that targets a member of the miR-125 family, serving as an independent prognostic marker for overall survival and disease-free survival in gastric cancer patients (37). These studies indicate that circRNA can be used as a potential diagnostic and prognostic marker for cancer in the future. However, to date, no study has been published on the dysregulation of circRNAs in UM.
We pioneered the study on the expression of circRNAs in UM by microarray profiling. We found 50,579 circRNAs with differential expression in five paired of UM and normal uvea tissues (FC≥2.0; P<0.05). Among them, 20,654 circRNAs were up-regulated and 29,925 circRNAs were down-regulated. Considering the false-positive rates of circRNA prediction tools (38), experimental methods are required to validate results of prediction tools and select high-confident circRNAs for further study (39). Thus, we randomly selected 15 differentially expressed circRNAs in the expression profile and used qRT-PCR to verify their expression levels in 20 UM tissues and 20 normal uvea tissues. The results showed that the alteration of seven circRNAs identified by microarray profiling — hsa_circ_0119873, hsa_circ_0128533, hsa_circ_0047924, hsa_circ_0103232, hsa-circRNA10628-6, hsa_circ_0032148 and hsa_circ_0133460 — was confirmed by the scale-up validation. More interestingly, the most significant validated up-regulated circRNA, hsa_circ_0119873, is predicted to target the Ras guanine nucleotide-releasing protein 3 gene (RasGRP3). RasGRP3 plays an important role in UM, and some studies have indicated it acts as a potential therapeutic target for UM driven by oncogenic GNAQ/11 mutation (40,41). The diacylglycerol kinase iota gene (DGKI), a predicted target gene of hsa_circ_0133460, has been demonstrated to bind to RasGRP3 and affect Ras signaling pathway (42).
We then explored the potential biological functions and mechanisms of dysregulated circRNAs in UM by analyzing the GO and pathway enrichment analyses of their gene symbols. GO enrichment analysis indicated that some circRNAs may be involved in the regulation of critical biological processes, cellular components and molecular functions. Particularly, according to the pathway analysis, some circRNAs play roles in the PI3K/Akt signaling pathway as well as the Wnt signaling pathway. The PI3K/Akt pathway is highly active in most UM cases, and elevated levels of AKT phosphorylation increase the risk of UM metastasis (43,44), while inhibition of the PI3K/Akt pathway can lead to tumor cell cycle arrest and reduced growth (45). Studies have found an increase in Wnt5a in primary UM that is associated with a poor prognosis in patients. It was also found that blocking the Wnt signaling pathway can inhibit the growth, migration and invasion of UM cells (46). These studies indicate that circRNAs may be involved in the occurrence and development of UM.
Many studies have shown that circRNAs can act as miRNA sponges or as potent ceRNA molecules (47-49). Because miRNAs play important roles in the progression of UM (50), some circRNAs may participate in UM by interacting with miRNAs. Using miRNA target prediction software, the circRNA-miRNA-mRNA regulatory networks were predicted, facilitating the understanding of the potential roles of the validated circRNAs. The bioinformatics analysis indicated that most circRNAs contained one or more miRNA binding sites. The association of miRNAs with UM suggests that circRNAs may play regulatory roles in UM. It is worth noting that the overexpression of miR-145 can inhibit the proliferation of UM cells and promote UM cell apoptosis (51). However, hsa_circ_0128533 can be predicted to bind miR-145; thus, hsa_circ_0128533 may be an oncogene and prevent UM apoptosis by targeting miR-145. Moreover, our analysis suggests that the up-regulated hsa_circ_0119873 may target hsa-miR-92a-3p, hsa-miR-193a, hsa-miR-193b, and hsa-miR-204 (Supplementary Figure S1). Down-regulation of miR-92 in breast epithelial cells enhances the invasion of breast cancer, suggesting that this may be a potential therapeutic target for breast cancer (52). miR-193 also exerts direct effects on the proliferation, migration and invasion of human intrahepatic cholangiocarcinoma cells by regulating TGFBR3 (53). Overexpressing miR-204-5p suppressed papillary thyroid carcinoma cell proliferation and induced cell cycle arrest and apoptosis (54). Our results suggest that it is worthwhile to further study these new dysregulated circRNAs as miRNA sponges and their potential biological functions in the development of UM.
Conclusions
Our study revealed, for the first time, that circRNAs are dysregulated in UM tissues compared with normal choroid tissues. For differentially expressed circRNAs, we performed GO analysis and pathway analysis, and we analyzed several cancer-associated circRNA-miRNA axes that may be involved in the pathogenesis of UM. We verified the expression levels of seven dysregulated circRNAs in 20 UM samples and 20 normal uvea samples by qRT-PCR that were consistent with the microarray data. Our results would be helpful for future studies of circRNAs in UM; however more experiments with more samples are needed to explain the underlying molecular mechanisms of circRNAs in UM.
Acknowledgements
This study was supported by Beijing Municipal Administration of Hospitals’ Ascent Plan (No. DFL20150201), the National Natural Science Foundation of China (No. 81570891), Beijing Natural Science Foundation (No. 7151003), Advanced Health Care Professionals Development Project of Beijing Municipal Health Bureau (No. 2014-2-003) and The Capital Health Research and Development of Special (No. 2016-1-2051).
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Krantz BA, Dave N, Komatsubara KM, et al. Uveal melanoma: epidemiology, etiology, and treatment of primary disease. Clin Ophthalmol 2017;11:279–89. [PubMed] DOI:10.2147/OPTH.S89591
- Shields CL, Kaliki S, Furuta M, et al. American Joint Committee on Cancer Classification of Uveal Melanoma (Anatomic Stage) Predicts Prognosis in 7,731 Patients: The 2013 Zimmerman Lecture. Ophthalmology 2015;122:1180–6. [PubMed] DOI:10.1016/j.ophtha.2015.01.026
- Shields JA, Shields CL. Management of posterior uveal melanoma: past, present, and future: the 2014 Charles L. Schepens lecture. Ophthalmology 2015;122:414–28. [PubMed] DOI:10.1016/j.ophtha.2014.08.046
- Eskelin S, Pyrhönen S, Summanen P, et al. Tumor doubling times in metastatic malignant melanoma of the uvea: tumor progression before and after treatment. Ophthalmology 2000;107:1443–9. [PubMed] DOI:10.1016/S0161-6420(00)00182-2
- Kujala E, Mäkitie T, Kivelä T. Very long-term prognosis of patients with malignant uveal melanoma. Invest Ophthalmol Vis Sci 2003;44:4651–9. [PubMed] DOI:10.1167/iovs.03-0538
- Triozzi PL, Singh AD. Adjuvant therapy of uveal melanoma: current status. Ocul Oncol Pathol 2014;1:54–62. DOI:10.1159/000367715
- Shields CL, Furuta M, Thangappan A, et al. Metastasis of uveal melanoma millimeter-by-millimeter in 8033 consecutive eyes. Arch Ophthalmol 2009;127:989–98. [PubMed] DOI:10.1001/archophthalmol.2009.208
- Qu S, Yang X, Li X, et al. Circular RNA: A new star of noncoding RNAs. Cancer Lett 2015;365:141–8. DOI:10.1016/j.canlet.2015.06.003
- Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013;19:141–57. [PubMed] DOI:10.1261/rna.035667.112
- Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013;495:333–8. [PubMed] DOI:10.1038/nature11928
- Chen LL, Yang L. Regulation of circRNA biogenesis. RNA Biol 2015;12:381–8. [PubMed] DOI:10.1080/15476286.2015.1020271
- Hentze MW, Preiss T. Circular RNAs: splicing’s enigma variations. EMBO J 2013;32:923–5. [PubMed] DOI:10.1038/emboj.2013.53
- Ashwal-Fluss R, Meyer M, Pamudurti NR, et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 2014;56:55–66. [PubMed] DOI:10.1016/j.molcel.2014.08.019
- Starke S, Jost I, Rossbach O, et al. Exon circularization requires canonical splice signals. Cell Rep 2015;10:103–11. [PubMed] DOI:10.1016/j.celrep.2014.12.002
- Chen X, Fan S, Song E. Noncoding RNAs: New Players in Cancers. Adv Exp Med Biol 2016;927:1–47. [PubMed] DOI:10.1007/978-981-10-1498-7_1
- Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87–108. [PubMed] DOI:10.3322/caac.21262
- Yang L, Li N, Wang C, et al. Cyclin L2, a novel RNA polymerase II-associated cyclin, is involved in pre-mRNA splicing and induces apoptosis of human hepatocellular carcinoma cells. J Biol Chem 2004;279:11639–48. [PubMed] DOI:10.1074/jbc.M312895200
- Suzuki H, Zuo Y, Wang J, et al. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res 2006;34:e63. [PubMed] DOI:10.1093/nar/gkl151
- Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature 2013;495:384–8. [PubMed] DOI:10.1038/nature11993
- Piwecka M, Glažar P, Hernandez-Miranda LR, et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 2017;357:Pii:eaam8526. DOI:10.1126/science.aam8526
- Worley LA, Long MD, Onken MD, et al. Micro-RNAs associated with metastasis in uveal melanoma identified by multiplexed microarray profiling. Melanoma Res 2008;18:184–90. [PubMed] DOI:10.1097/CMR.0b013e3282feeac6
- Radhakrishnan A, Badhrinarayanan N, Biswas J, et al. Analysis of chromosomal aberration (1, 3, and 8) and association of microRNAs in uveal melanoma. Mol Vis 2009;15:2146–54. [PubMed]
- Dong F, Lou D. MicroRNA-34b/c suppresses uveal melanoma cell proliferation and migration through multiple targets. Mol Vis 2012;18:537–46.
- Zhou J, Wan J, Gao X, et al. Dynamic m6A mRNA methylation directs translational control of heat shock response . Nature 2015;526:591–4. [PubMed] DOI:10.1038/nature15377
- Wang X, Zhao BS, Roundtree IA, et al. N6-methyladenosine modulates messenger RNA translation efficiency . Cell 2015;161:1388–99. [PubMed] DOI:10.1016/j.cell.2015.05.014
- Yang Y, Fan X, Mao M, et al. Extensive translation of circular RNAs driven by N6-methyladenosine . Cell Res 2017;27:626–41. DOI:10.1038/cr.2017.31
- Yang Y, Gao X, Zhang M, et al. Novel role of FBXW7 circular RNA in repressing glioma tumorigenesis. J Natl Cancer Inst 2018;110. DOI:10.1093/jnci/djx166
- Kristensen LS, Hansen TB, Venø MT, et al. Circular RNAs in cancer: opportunities and challenges in the field. Oncogene 2018;37:555–65. [PubMed] DOI:10.1038/onc.2017.361
- Rong D, Tang W, Li Z, et al. Novel insights into circular RNAs in clinical application of carcinomas. Onco Targets Ther 2017;10:2183–8. DOI:10.2147/OTT.S134403
- You X, Vlatkovic I, Babic A, et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci 2015;18:603–10. [PubMed] DOI:10.1038/nn.3975
- Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014;505:344–52. [PubMed] DOI:10.1038/nature12986
- Reichstein D. New concepts in the molecular understanding of uveal melanoma. Curr Opin Ophthalmol 2017;28:219–27. [PubMed] DOI:10.1097/ICU.0000000000000366
- Xing Y, Wen X, Ding X, et al. CANT1 lncRNA triggers efficient therapeutic efficacy by correcting aberrant lncing cascade in malignant uveal melanoma . Mol Ther 2017;25:1209–21. [PubMed] DOI:10.1016/j.ymthe.2017.02.016
- Qu S, Zhong Y, Shang R, et al. The emerging landscape of circular RNA in life processes. RNA Biol 2017;14:992–9. [PubMed] DOI:10.1080/15476286.2016.1220473
- Lyu D, Huang S. The emerging role and clinical implication of human exonic circular RNA. RNA Biol 2017;14:1000–6. [PubMed] DOI:10.1080/15476286.2016.1227904
- Han D, Li J, Wang H, et al. Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatology 2017;66:1151–64. DOI:10.1002/hep.29270
- Chen J, Li Y, Zheng Q, et al. Circular RNA profile identifies circPVT1 as a proliferative factor and prognostic marker in gastric cancer. Cancer Lett 2017;388:208–19. [PubMed] DOI:10.1016/j.canlet.2016.12.006
- Li S, Teng S, Xu J, et al. Microarray is an efficient tool for circRNA profiling. Brief Bioinform 2018. [Epub ahead of print]
- Fang Y, Wang X, Li W, et al. Screening of circular RNAs and validation of circANKRD36 associated with inflammation in patients with type 2 diabetes mellitus. Int J Mol Med 2018;42:1865–74. [PubMed] DOI:10.3892/ijmm.2018.3783
- Chen X, Wu Q, Depeille P, et al. RasGRP3 mediates MAPK pathway activation in GNAQ mutant uveal melanoma. Cancer Cell 2017;31:685–96. e6. [PubMed] DOI:10.1016/j.ccell.2017.04.002
- Moore AR, Ran L, Guan Y, et al. GNA11 Q209L mouse model reveals RasGRP3 as an essential signaling node in uveal melanoma. Cell Rep 2018;22:2455–68. [PubMed] DOI:10.1016/j.celrep.2018.01.081
- Regier DS, Higbee J, Lund KM, et al. Diacylglycerol kinase iota regulates Ras guanyl-releasing protein 3 and inhibits Rap1 signaling. Proc Natl Acad Sci U S A 2005;102:7595–600. [PubMed] DOI:10.1073/pnas.0500663102
- Pópulo H, Soares P, Rocha AS, et al. Evaluation of the mTOR pathway in ocular (uvea and conjunctiva) melanoma. Melanoma Res 2010;20:107–17. [PubMed] DOI:10.1097/CMR.0b013e32832ccd09
- Saraiva VS, Caissie AL, Segal L, et al. Immunohistochemical expression of phospho-Akt in uveal melanoma. Melanoma Res 2005;15:245–50. [PubMed] DOI:10.1097/00008390-200508000-00003
- Khalili JS, Yu X, Wang J, et al. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin Cancer Res 2012;18:4345–55. DOI:10.1158/1078-0432.CCR-11-3227
- Zuidervaart W, Pavey S, van Nieuwpoort FA, et al. Expression of Wnt5a and its downstream effector beta-catenin in uveal melanoma. Melanoma Res 2007;17:380–6. [PubMed] DOI:10.1097/CMR.0b013e3282f1d302
- Zheng Q, Bao C, Guo W, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun 2016;7:11215. [PubMed] DOI:10.1038/ncomms11215
- Li Y, Zheng F, Xiao X, et al. CircHIPK3 sponges miR-558 to suppress heparanase expression in bladder cancer cells. EMBO Rep 2017;18:1646–59. [PubMed] DOI:10.15252/embr.201643581
- Shan K, Liu C, Liu BH, et al. Circular noncoding RNA HIPK3 mediates retinal vascular dysfunction in diabetes mellitus. Circulation 2017;136:1629–42. [PubMed] DOI:10.1161/CIRCULATIONAHA.117.029004
- Amaro A, Gangemi R, Piaggio F, et al. The biology of uveal melanoma. Cancer Metastasis Rev 2017;36:109–40. [PubMed] DOI:10.1007/s10555-017-9663-3
- Li Y, Huang Q, Shi X, et al. MicroRNA 145 may play an important role in uveal melanoma cell growth by potentially targeting insulin receptor substrate-1. Chin Med J (Engl) 2014;127:1410–6. [PubMed]
- Smith L, Baxter EW, Chambers PA, et al. Down-regulation of miR-92 in breast epithelial cells and in normal but not tumour fibroblasts contributes to breast carcinogenesis. PLoS One 2015;10:e0139698. DOI:10.1371/journal.pone.0139698
- Han YL, Yin JJ, Cong JJ. Downregulation of microRNA-193-3p inhibits the progression of intrahepatic cholangiocarcinoma cells by upregulating TGFBR3. Exp Ther Med 2018;15:4508–14. [PubMed] DOI:10.3892/etm.2018.5958
- Liu L, Wang J, Li X, et al. MiR-204-5p suppresses cell proliferation by inhibiting IGFBP5 in papillary thyroid carcinoma. Biochem Biophys Res Commun 2015;457:621–6. [PubMed] DOI:10.1016/j.bbrc.2015.01.037